├── vignettes
├── .gitignore
└── cytotalk_guide.Rmd
├── LICENSE
├── .gitignore
├── data
├── lrp_human.rda
├── lrp_mouse.rda
├── pcg_human.rda
├── pcg_mouse.rda
├── protetta.rda
├── scrna_cpdb.rda
├── scrna_cyto.rda
└── result_cyto.rda
├── tests
├── testthat.R
└── testthat
│ └── test_matrix.R
├── docs
├── cytotalk_diagram.png
└── pathway.svg
├── .Rbuildignore
├── man
├── protetta.Rd
├── scrna_cyto.Rd
├── result_cyto.Rd
├── scrna_cpdb.Rd
├── pcg_human.Rd
├── pcg_mouse.Rd
├── lrp_human.Rd
├── lrp_mouse.Rd
├── from_single_cell_experiment.Rd
├── group_meta.Rd
├── ungroup_meta.Rd
├── doc_parallel.Rd
├── subset_rownames.Rd
├── extract_group.Rd
├── check_count_data.Rd
├── normalize_sparse.Rd
├── subset_non_zero.Rd
├── match_lr_pairs.Rd
├── read_matrix_folder.Rd
├── doc_pem.Rd
├── read_matrix_with_meta.Rd
├── doc_graphing.Rd
├── doc_mutinfo.Rd
├── analyze_pathway.Rd
├── doc_pcst.Rd
├── doc_integrated.Rd
└── run_cytotalk.Rd
├── R
├── parallel.R
├── utils.R
├── pem.R
├── mutinfo.R
├── fileio.R
├── analysis.R
├── graphing.R
├── matrix.R
├── integrated.R
├── data.R
├── pcst.R
└── cytotalk.R
├── NAMESPACE
├── LICENSE.md
├── DESCRIPTION
├── README.md
└── README.Rmd
/vignettes/.gitignore:
--------------------------------------------------------------------------------
1 | *.html
2 | *.R
3 |
--------------------------------------------------------------------------------
/LICENSE:
--------------------------------------------------------------------------------
1 | YEAR: 2021
2 | COPYRIGHT HOLDER: CytoTalk authors
3 |
--------------------------------------------------------------------------------
/.gitignore:
--------------------------------------------------------------------------------
1 | .Rproj.user
2 | .Rhistory
3 | inst/doc
4 | test_script.R*
5 | /doc/
6 | /Meta/
7 |
--------------------------------------------------------------------------------
/data/lrp_human.rda:
--------------------------------------------------------------------------------
https://raw.githubusercontent.com/tanlabcode/CytoTalk/HEAD/data/lrp_human.rda
--------------------------------------------------------------------------------
/data/lrp_mouse.rda:
--------------------------------------------------------------------------------
https://raw.githubusercontent.com/tanlabcode/CytoTalk/HEAD/data/lrp_mouse.rda
--------------------------------------------------------------------------------
/data/pcg_human.rda:
--------------------------------------------------------------------------------
https://raw.githubusercontent.com/tanlabcode/CytoTalk/HEAD/data/pcg_human.rda
--------------------------------------------------------------------------------
/data/pcg_mouse.rda:
--------------------------------------------------------------------------------
https://raw.githubusercontent.com/tanlabcode/CytoTalk/HEAD/data/pcg_mouse.rda
--------------------------------------------------------------------------------
/data/protetta.rda:
--------------------------------------------------------------------------------
https://raw.githubusercontent.com/tanlabcode/CytoTalk/HEAD/data/protetta.rda
--------------------------------------------------------------------------------
/data/scrna_cpdb.rda:
--------------------------------------------------------------------------------
https://raw.githubusercontent.com/tanlabcode/CytoTalk/HEAD/data/scrna_cpdb.rda
--------------------------------------------------------------------------------
/data/scrna_cyto.rda:
--------------------------------------------------------------------------------
https://raw.githubusercontent.com/tanlabcode/CytoTalk/HEAD/data/scrna_cyto.rda
--------------------------------------------------------------------------------
/tests/testthat.R:
--------------------------------------------------------------------------------
1 | library(testthat)
2 | library(CytoTalk)
3 |
4 | test_check("CytoTalk")
5 |
--------------------------------------------------------------------------------
/data/result_cyto.rda:
--------------------------------------------------------------------------------
https://raw.githubusercontent.com/tanlabcode/CytoTalk/HEAD/data/result_cyto.rda
--------------------------------------------------------------------------------
/docs/cytotalk_diagram.png:
--------------------------------------------------------------------------------
https://raw.githubusercontent.com/tanlabcode/CytoTalk/HEAD/docs/cytotalk_diagram.png
--------------------------------------------------------------------------------
/.Rbuildignore:
--------------------------------------------------------------------------------
1 | ^CytoTalk\.Rproj$
2 | ^\.Rproj\.user$
3 |
4 | ^docs$
5 | ^doc$
6 | ^Meta$
7 |
8 | ^README\.Rmd$
9 | ^LICENSE\.md$
10 | ^test_script\.R$
11 | ^test_script\.Rmd$
12 |
--------------------------------------------------------------------------------
/man/protetta.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/data.R
3 | \docType{data}
4 | \name{protetta}
5 | \alias{protetta}
6 | \title{Protetta Stone}
7 | \format{
8 | A dataframe
9 | }
10 | \usage{
11 | protetta
12 | }
13 | \description{
14 | Protetta Stone
15 | }
16 | \keyword{datasets}
17 |
--------------------------------------------------------------------------------
/man/scrna_cyto.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/data.R
3 | \docType{data}
4 | \name{scrna_cyto}
5 | \alias{scrna_cyto}
6 | \title{Example scRNAseq Data (CytoTalk)}
7 | \format{
8 | A list
9 | }
10 | \usage{
11 | scrna_cyto
12 | }
13 | \description{
14 | Example scRNAseq Data (CytoTalk)
15 | }
16 | \keyword{datasets}
17 |
--------------------------------------------------------------------------------
/man/result_cyto.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/data.R
3 | \docType{data}
4 | \name{result_cyto}
5 | \alias{result_cyto}
6 | \title{Example Pipeline Result (CytoTalk)}
7 | \format{
8 | A list
9 | }
10 | \usage{
11 | result_cyto
12 | }
13 | \description{
14 | Example Pipeline Result (CytoTalk)
15 | }
16 | \keyword{datasets}
17 |
--------------------------------------------------------------------------------
/man/scrna_cpdb.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/data.R
3 | \docType{data}
4 | \name{scrna_cpdb}
5 | \alias{scrna_cpdb}
6 | \title{Example scRNAseq Data (CellphoneDB)}
7 | \format{
8 | A list
9 | }
10 | \usage{
11 | scrna_cpdb
12 | }
13 | \description{
14 | Example scRNAseq Data (CellphoneDB)
15 | }
16 | \keyword{datasets}
17 |
--------------------------------------------------------------------------------
/man/pcg_human.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/data.R
3 | \docType{data}
4 | \name{pcg_human}
5 | \alias{pcg_human}
6 | \title{Human protein-coding genes (PCG)}
7 | \format{
8 | A large character vector
9 | }
10 | \usage{
11 | pcg_human
12 | }
13 | \description{
14 | Contains the names of human protein-coding genes
15 | }
16 | \keyword{datasets}
17 |
--------------------------------------------------------------------------------
/man/pcg_mouse.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/data.R
3 | \docType{data}
4 | \name{pcg_mouse}
5 | \alias{pcg_mouse}
6 | \title{Mouse protein-coding genes (PCG)}
7 | \format{
8 | A large character vector
9 | }
10 | \usage{
11 | pcg_mouse
12 | }
13 | \description{
14 | Contains the names of mouse protein-coding genes
15 | }
16 | \keyword{datasets}
17 |
--------------------------------------------------------------------------------
/tests/testthat/test_matrix.R:
--------------------------------------------------------------------------------
1 | test_that("grouping is identical to extraction", {
2 | c <- 6
3 |
4 | mat <- matrix(runif(10 * c), ncol = c)
5 | meta <- paste(as.integer((seq_len(c) - 1) / 2))
6 |
7 | lst <- CytoTalk:::new_named_list(mat, meta)
8 |
9 | expect_identical(
10 | CytoTalk::extract_group(meta[1], lst),
11 | CytoTalk::group_meta(lst)[[meta[1]]]
12 | )
13 | })
14 |
--------------------------------------------------------------------------------
/man/lrp_human.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/data.R
3 | \docType{data}
4 | \name{lrp_human}
5 | \alias{lrp_human}
6 | \title{Human ligand-receptor pairs}
7 | \format{
8 | A data frame with two variables: \code{ligand} and \code{receptor}
9 | }
10 | \usage{
11 | lrp_human
12 | }
13 | \description{
14 | Contains the names of human ligand-receptor pairs
15 | }
16 | \keyword{datasets}
17 |
--------------------------------------------------------------------------------
/man/lrp_mouse.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/data.R
3 | \docType{data}
4 | \name{lrp_mouse}
5 | \alias{lrp_mouse}
6 | \title{Mouse ligand-receptor pairs}
7 | \format{
8 | A data frame with two variables: \code{ligand} and \code{receptor}
9 | }
10 | \usage{
11 | lrp_mouse
12 | }
13 | \description{
14 | Contains the names of mouse ligand-receptor pairs
15 | }
16 | \keyword{datasets}
17 |
--------------------------------------------------------------------------------
/R/parallel.R:
--------------------------------------------------------------------------------
1 | #' @rdname doc_parallel
2 | #' @export
3 | unregister_parallel <- function() {
4 | doParallel::stopImplicitCluster()
5 | }
6 |
7 | #' @rdname doc_parallel
8 | #' @export
9 | register_parallel <- function(cores=NULL) {
10 | if (is.null(cores)) {
11 | cores <- parallel::detectCores()
12 | }
13 | cores <- max(1, cores)
14 | if (cores != 1) {
15 | doParallel::registerDoParallel(cores = cores)
16 | }
17 | }
18 |
--------------------------------------------------------------------------------
/man/from_single_cell_experiment.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/fileio.R
3 | \name{from_single_cell_experiment}
4 | \alias{from_single_cell_experiment}
5 | \title{Convert SingleCellExperiment to Named List}
6 | \usage{
7 | from_single_cell_experiment(sce)
8 | }
9 | \arguments{
10 | \item{sce}{SingleCellExperiment object}
11 | }
12 | \value{
13 | A named list containing a sparse data matrix and cell type metadata
14 | }
15 | \description{
16 | Convert SingleCellExperiment to Named List
17 | }
18 |
--------------------------------------------------------------------------------
/man/group_meta.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/matrix.R
3 | \name{group_meta}
4 | \alias{group_meta}
5 | \title{Meta List to Named List}
6 | \usage{
7 | group_meta(lst)
8 | }
9 | \arguments{
10 | \item{lst}{A meta matrix list (outputted from \code{doc_fileio} methods)}
11 | }
12 | \value{
13 | A list of matrices
14 | }
15 | \description{
16 | Meta List to Named List
17 | }
18 | \examples{
19 | {
20 | lst_scrna <- CytoTalk::scrna_cyto
21 | result <- group_meta(lst_scrna)
22 | }
23 |
24 | }
25 |
--------------------------------------------------------------------------------
/man/ungroup_meta.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/matrix.R
3 | \name{ungroup_meta}
4 | \alias{ungroup_meta}
5 | \title{Named List to Meta List}
6 | \usage{
7 | ungroup_meta(lst)
8 | }
9 | \arguments{
10 | \item{lst}{A meta matrix list (outputted from \code{doc_fileio} methods)}
11 | }
12 | \value{
13 | A list of a matrix and a meta vector
14 | }
15 | \description{
16 | Named List to Meta List
17 | }
18 | \examples{
19 | {
20 | lst_scrna <- CytoTalk::scrna_cyto
21 | lst_group <- group_meta(lst_scrna)
22 | result <- ungroup_meta(lst_group)
23 | }
24 |
25 | }
26 |
--------------------------------------------------------------------------------
/man/doc_parallel.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/data.R, R/parallel.R
3 | \name{doc_parallel}
4 | \alias{doc_parallel}
5 | \alias{unregister_parallel}
6 | \alias{register_parallel}
7 | \title{Registering Parallel Backend}
8 | \usage{
9 | unregister_parallel()
10 |
11 | register_parallel(cores = NULL)
12 | }
13 | \arguments{
14 | \item{cores}{How many cores to use for parallel processing?}
15 | }
16 | \value{
17 | Nothing
18 | }
19 | \description{
20 | Registering Parallel Backend
21 | }
22 | \examples{
23 | {
24 | register_parallel(cores=2)
25 | unregister_parallel()
26 | }
27 |
28 | }
29 |
--------------------------------------------------------------------------------
/man/subset_rownames.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/matrix.R
3 | \name{subset_rownames}
4 | \alias{subset_rownames}
5 | \title{Subset Rows on Rownames}
6 | \usage{
7 | subset_rownames(mat, labels)
8 | }
9 | \arguments{
10 | \item{mat}{A numerical matrix}
11 |
12 | \item{labels}{A subset of the matrix's rownames to filter by}
13 | }
14 | \value{
15 | A matrix
16 | }
17 | \description{
18 | Subset Rows on Rownames
19 | }
20 | \examples{
21 | {
22 | lst_scrna <- CytoTalk::scrna_cyto
23 | pcg <- CytoTalk::pcg_human
24 | cell_type_a <- "Macrophages"
25 | mat_a <- extract_group(cell_type_a, lst_scrna)
26 | result <- subset_rownames(mat_a, pcg)
27 | }
28 |
29 | }
30 |
--------------------------------------------------------------------------------
/man/extract_group.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/matrix.R
3 | \name{extract_group}
4 | \alias{extract_group}
5 | \title{Extract Cell Type from Meta List}
6 | \usage{
7 | extract_group(group, lst)
8 | }
9 | \arguments{
10 | \item{group}{Character vector with a single string, name of cell type to
11 | extract}
12 |
13 | \item{lst}{A meta matrix list (outputted from \code{doc_fileio} methods)}
14 | }
15 | \value{
16 | A matrix
17 | }
18 | \description{
19 | Extract Cell Type from Meta List
20 | }
21 | \examples{
22 | {
23 | lst_scrna <- CytoTalk::scrna_cyto
24 | cell_type_a <- "Macrophages"
25 | result <- extract_group(cell_type_a, lst_scrna)
26 | }
27 |
28 | }
29 |
--------------------------------------------------------------------------------
/man/check_count_data.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/matrix.R
3 | \name{check_count_data}
4 | \alias{check_count_data}
5 | \title{Transform Count Data if Detected}
6 | \usage{
7 | check_count_data(mat, auto_transform = TRUE)
8 | }
9 | \arguments{
10 | \item{mat}{A numerical matrix}
11 |
12 | \item{auto_transform}{Should the data be transformed if counts are detected?}
13 | }
14 | \value{
15 | A matrix
16 | }
17 | \description{
18 | Transform Count Data if Detected
19 | }
20 | \examples{
21 | {
22 | lst_scrna <- CytoTalk::scrna_cyto
23 | cell_type_a <- "Macrophages"
24 | mat_a <- extract_group(cell_type_a, lst_scrna)
25 | result <- check_count_data(mat_a)
26 | }
27 |
28 | }
29 |
--------------------------------------------------------------------------------
/man/normalize_sparse.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/matrix.R
3 | \name{normalize_sparse}
4 | \alias{normalize_sparse}
5 | \title{scRNAseq Count Normalization}
6 | \usage{
7 | normalize_sparse(mat, scale.factor = 10000)
8 | }
9 | \arguments{
10 | \item{mat}{An integer matrix}
11 |
12 | \item{scale.factor}{A single number constant by which to scale the
13 | transformed data by}
14 | }
15 | \value{
16 | A matrix
17 | }
18 | \description{
19 | scRNAseq Count Normalization
20 | }
21 | \examples{
22 | {
23 | lst_scrna <- CytoTalk::scrna_cyto
24 | cell_type_a <- "Macrophages"
25 | mat_a <- extract_group(cell_type_a, lst_scrna)
26 | result <- normalize_sparse(mat_a)
27 | }
28 |
29 | }
30 |
--------------------------------------------------------------------------------
/man/subset_non_zero.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/matrix.R
3 | \name{subset_non_zero}
4 | \alias{subset_non_zero}
5 | \title{Subset Rows on Proportion Non-Zero}
6 | \usage{
7 | subset_non_zero(mat, cutoff)
8 | }
9 | \arguments{
10 | \item{mat}{A numerical matrix}
11 |
12 | \item{cutoff}{Threshold value in range [0, 1]; proportion of each row
13 | required to be non-zero}
14 | }
15 | \value{
16 | A matrix
17 | }
18 | \description{
19 | Subset Rows on Proportion Non-Zero
20 | }
21 | \examples{
22 | {
23 | lst_scrna <- CytoTalk::scrna_cyto
24 | cell_type_a <- "Macrophages"
25 | cutoff_a <- 0.8
26 | mat_a <- extract_group(cell_type_a, lst_scrna)
27 | result <- subset_non_zero(mat_a, cutoff_a)
28 | }
29 |
30 | }
31 |
--------------------------------------------------------------------------------
/man/match_lr_pairs.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/matrix.R
3 | \name{match_lr_pairs}
4 | \alias{match_lr_pairs}
5 | \title{Find Ligand Receptor Pairs in Matrix}
6 | \usage{
7 | match_lr_pairs(mat, lrp)
8 | }
9 | \arguments{
10 | \item{mat}{A numerical matrix}
11 |
12 | \item{lrp}{Ligand-receptor pair data.frame; see \code{CytoTalk::lrp_human} for
13 | an example}
14 | }
15 | \value{
16 | A matrix with two columns (ligand-receptor)
17 | }
18 | \description{
19 | Find Ligand Receptor Pairs in Matrix
20 | }
21 | \examples{
22 | {
23 | lst_scrna <- CytoTalk::scrna_cyto
24 | lrp <- CytoTalk::lrp_human
25 | cell_type_a <- "Macrophages"
26 | mat_a <- extract_group(cell_type_a, lst_scrna)
27 | result <- match_lr_pairs(mat_a, lrp)
28 | }
29 |
30 | }
31 |
--------------------------------------------------------------------------------
/NAMESPACE:
--------------------------------------------------------------------------------
1 | # Generated by roxygen2: do not edit by hand
2 |
3 | export(analyze_pathway)
4 | export(check_count_data)
5 | export(crosstalk)
6 | export(discretize_sparse)
7 | export(extract_group)
8 | export(extract_network)
9 | export(extract_pathways)
10 | export(from_single_cell_experiment)
11 | export(gene_relevance)
12 | export(graph_pathway)
13 | export(group_meta)
14 | export(integrate_network)
15 | export(ks_test_pcst)
16 | export(match_lr_pairs)
17 | export(mi_mat_parallel)
18 | export(mutinfo_xy)
19 | export(node_prize)
20 | export(nonselftalk)
21 | export(normalize_sparse)
22 | export(pem)
23 | export(pem_basic)
24 | export(read_matrix_folder)
25 | export(read_matrix_with_meta)
26 | export(register_parallel)
27 | export(run_cytotalk)
28 | export(run_pcst)
29 | export(subset_non_zero)
30 | export(subset_rownames)
31 | export(summarize_pcst)
32 | export(ungroup_meta)
33 | export(unregister_parallel)
34 | export(write_network_sif)
35 |
--------------------------------------------------------------------------------
/man/read_matrix_folder.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/fileio.R
3 | \name{read_matrix_folder}
4 | \alias{read_matrix_folder}
5 | \title{Read Folder with scRNAseq Data}
6 | \usage{
7 | read_matrix_folder(
8 | dpath,
9 | pattern = ".*scRNAseq_(.+?)\\\\..+",
10 | auto_transform = TRUE
11 | )
12 | }
13 | \arguments{
14 | \item{dpath}{The path of a directory, which contains scRNAseq matrices}
15 |
16 | \item{pattern}{A regular expression, matches scRNAseq filenames}
17 |
18 | \item{auto_transform}{Should count data be transformed if detected?}
19 | }
20 | \value{
21 | A named list containing a sparse data matrix and cell type metadata
22 | }
23 | \description{
24 | Read Folder with scRNAseq Data
25 | }
26 | \examples{
27 | {
28 | dir_in <- "~/Tan-Lab/scRNAseq-data"
29 | # lst_scrna <- CytoTalk::read_matrix_folder(dir_in)
30 | # result <- str(lst_scrna$cell_types)
31 | }
32 |
33 | }
34 |
--------------------------------------------------------------------------------
/man/doc_pem.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/data.R, R/pem.R
3 | \name{doc_pem}
4 | \alias{doc_pem}
5 | \alias{pem_basic}
6 | \alias{pem}
7 | \title{Preferential Expression Measure}
8 | \usage{
9 | pem_basic(mat_scrna, labels)
10 |
11 | pem(lst_scrna)
12 | }
13 | \arguments{
14 | \item{mat_scrna}{Matrix containing scRNA-seq data, along with \code{labels},
15 | defines the transformed count data and the associated cell types}
16 |
17 | \item{labels}{Associated cell types, column-wise, of \code{mat_scrna}}
18 |
19 | \item{lst_scrna}{List containing scRNA-seq data; for example, lists returned
20 | from \code{read_matrix_folder} or \code{read_matrix_with_meta}}
21 | }
22 | \value{
23 | Preferential expression measure scores
24 | }
25 | \description{
26 | Preferential Expression Measure
27 | }
28 | \examples{
29 | {
30 | lst_scrna <- CytoTalk::scrna_cyto
31 | result <- pem(lst_scrna)
32 | }
33 |
34 | }
35 |
--------------------------------------------------------------------------------
/man/read_matrix_with_meta.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/fileio.R
3 | \name{read_matrix_with_meta}
4 | \alias{read_matrix_with_meta}
5 | \title{Read scRNAseq Data Matrix and Metadata}
6 | \usage{
7 | read_matrix_with_meta(fpath_mat, fpath_meta, auto_transform = TRUE)
8 | }
9 | \arguments{
10 | \item{fpath_mat}{The path of a file containing a scRNAseq data matrix}
11 |
12 | \item{fpath_meta}{The path of a file contianing column metadata (cell types)}
13 |
14 | \item{auto_transform}{Should count data be transformed if detected?}
15 | }
16 | \value{
17 | A named list containing a sparse data matrix and cell type metadata
18 | }
19 | \description{
20 | Read scRNAseq Data Matrix and Metadata
21 | }
22 | \examples{
23 | {
24 | fpath_mat <- "~/Tan-Lab/scRNAseq-data-cpdb/sample_counts.txt"
25 | fpath_meta <- "~/Tan-Lab/scRNAseq-data-cpdb/sample_meta.txt"
26 | # lst_scrna <- CytoTalk::read_matrix_with_meta(fpath_mat, fpath_meta)
27 | # result <- str(lst_scrna$cell_types)
28 | }
29 |
30 | }
31 |
--------------------------------------------------------------------------------
/man/doc_graphing.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/data.R, R/graphing.R
3 | \name{doc_graphing}
4 | \alias{doc_graphing}
5 | \alias{write_network_sif}
6 | \alias{graph_pathway}
7 | \title{Graphing with Cytoscape and Graphviz}
8 | \usage{
9 | write_network_sif(df_net, cell_type_a, dir_out)
10 |
11 | graph_pathway(df_net_sub)
12 | }
13 | \arguments{
14 | \item{df_net}{Final network; for example, the output of the
15 | \code{extract_network} function}
16 |
17 | \item{cell_type_a}{Name of cell type A that matches scRNA-seq file; for
18 | example, \code{"Fibroblasts"}}
19 |
20 | \item{dir_out}{Folder used for output}
21 |
22 | \item{df_net_sub}{A subset of the final network (pathway); for example, the
23 | output of the \code{extract_pathways} function}
24 | }
25 | \value{
26 | Graphs which represent the subset pathways of the final network
27 | }
28 | \description{
29 | Graphing with Cytoscape and Graphviz
30 | }
31 | \examples{
32 | {
33 | pathways <- CytoTalk::result_cyto$pathways$raw
34 | result <- graph_pathway(pathways[[1]])
35 | }
36 |
37 | }
38 |
--------------------------------------------------------------------------------
/LICENSE.md:
--------------------------------------------------------------------------------
1 | # MIT License
2 |
3 | Copyright (c) 2021 CytoTalk authors
4 |
5 | Permission is hereby granted, free of charge, to any person obtaining a copy
6 | of this software and associated documentation files (the "Software"), to deal
7 | in the Software without restriction, including without limitation the rights
8 | to use, copy, modify, merge, publish, distribute, sublicense, and/or sell
9 | copies of the Software, and to permit persons to whom the Software is
10 | furnished to do so, subject to the following conditions:
11 |
12 | The above copyright notice and this permission notice shall be included in all
13 | copies or substantial portions of the Software.
14 |
15 | THE SOFTWARE IS PROVIDED "AS IS", WITHOUT WARRANTY OF ANY KIND, EXPRESS OR
16 | IMPLIED, INCLUDING BUT NOT LIMITED TO THE WARRANTIES OF MERCHANTABILITY,
17 | FITNESS FOR A PARTICULAR PURPOSE AND NONINFRINGEMENT. IN NO EVENT SHALL THE
18 | AUTHORS OR COPYRIGHT HOLDERS BE LIABLE FOR ANY CLAIM, DAMAGES OR OTHER
19 | LIABILITY, WHETHER IN AN ACTION OF CONTRACT, TORT OR OTHERWISE, ARISING FROM,
20 | OUT OF OR IN CONNECTION WITH THE SOFTWARE OR THE USE OR OTHER DEALINGS IN THE
21 | SOFTWARE.
22 |
--------------------------------------------------------------------------------
/R/utils.R:
--------------------------------------------------------------------------------
1 | #' @noRd
2 | cmatch <- function(x, y) {
3 | match(toupper(x), toupper(y))
4 | }
5 |
6 | #' @noRd
7 | all_identical <- function(lst) {
8 | all(vapply(lst[-1], FUN = identical, logical(1), lst[[1]]))
9 | }
10 |
11 | #' @noRd
12 | format_message <- function(msg) {
13 | sprintf("%s\n", trimws(msg))
14 | }
15 |
16 | #' @noRd
17 | errorifnot <- function(expr, msg) {
18 | if (!expr) stop(format_message(msg))
19 | }
20 |
21 | #' @noRd
22 | warnifnot <- function(expr, msg) {
23 | if (!expr) warning(format_message(msg))
24 | }
25 |
26 | #' @noRd
27 | is_integer <- function(mat) {
28 | identical(mat, round(mat))
29 | }
30 |
31 | #' @noRd
32 | zero_diag <- function(mat) {
33 | diag(mat) <- 0
34 | mat
35 | }
36 |
37 | #' @noRd
38 | dir_full <- function(folder, ...) {
39 | fnames <- dir(folder, ...)
40 | file.path(folder, fnames)
41 | }
42 |
43 | #' @noRd
44 | totitle <- function(x) {
45 | gsub("^([A-z])", "\\U\\1", tolower(x), perl = TRUE)
46 | }
47 |
48 | #' @noRd
49 | add_suffix <- function(x, type) {
50 | sprintf("%s__%s", totitle(x), type)
51 | }
52 |
53 | #' @noRd
54 | minmax <- function(x, ...) {
55 | (x - min(x, ...)) / (max(x, ...) - min(x, ...))
56 | }
57 |
58 | #' @noRd
59 | zero_na_neg <- function(x) {
60 | ifelse(x < 0 | is.na(x), 0, x)
61 | }
62 |
63 | #' @noRd
64 | now <- function() {
65 | format(Sys.time(), "%H:%M:%S")
66 | }
67 |
68 | #' @noRd
69 | tick <- function(step, msg) {
70 | cat("[", step, " / 8] (", now(), ") ", msg, "\n", sep = "")
71 | }
72 |
--------------------------------------------------------------------------------
/R/pem.R:
--------------------------------------------------------------------------------
1 | #' @rdname doc_pem
2 | #' @export
3 | pem_basic <- function(mat_scrna, labels) {
4 | # exponentiate the normalized data
5 | mat_scrna <- expm1(mat_scrna)
6 | # seperate out the cell types,
7 | # rowmeans per scRNA file
8 | lst_means <- lapply(group_meta_basic(mat_scrna, labels), Matrix::rowMeans)
9 | # sums of rowmeans, per scRNA file
10 | lst_sums <- lapply(lst_means, sum)
11 | # sums of rowmeans, per gene (combined scRNA files)
12 | vec_gene_sums <- rowSums(do.call(cbind, lst_means))
13 | # overall sum of rowmeans
14 | total_sum <- sum(vec_gene_sums)
15 |
16 | # for all cell_types
17 | mat_pem <- list()
18 | for (i in seq_len(length(lst_means))) {
19 | mat_pem[[i]] <- vector()
20 |
21 | # what proportion of this cell type's rowmean sum
22 | # accounts for the whole?
23 | cell_type_prop <- lst_sums[[i]] / total_sum
24 |
25 | # for all genes
26 | for (j in seq_len(length(vec_gene_sums))) {
27 |
28 | # scale gene sum to cell type proportion
29 | gene_prop <- vec_gene_sums[j] * cell_type_prop
30 | # scale cell type rowmean to gene proportion
31 | mat_pem[[i]][j] <- log10(lst_means[[i]][j] / gene_prop)
32 | }
33 | }
34 |
35 | # join columns to dataframe
36 | mat_pem <- as.data.frame(do.call(cbind, mat_pem))
37 |
38 | # extract valid type names, copy over rownames
39 | colnames(mat_pem) <- names(lst_means)
40 | rownames(mat_pem) <- rownames(mat_scrna)
41 |
42 | # return pem matrix
43 | as.matrix(mat_pem)
44 | }
45 |
46 | #' @rdname doc_pem
47 | #' @export
48 | pem <- function(lst_scrna) {
49 | pem_basic(lst_scrna[[1]], lst_scrna[[2]])
50 | }
51 |
--------------------------------------------------------------------------------
/man/doc_mutinfo.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/data.R, R/mutinfo.R
3 | \name{doc_mutinfo}
4 | \alias{doc_mutinfo}
5 | \alias{discretize_sparse}
6 | \alias{mutinfo_xy}
7 | \alias{mi_mat_parallel}
8 | \title{Information Theory}
9 | \usage{
10 | discretize_sparse(mat, disc = "equalfreq", nbins = nrow(mat)^(1/3))
11 |
12 | mutinfo_xy(x, y, method = "emp", normalize = FALSE)
13 |
14 | mi_mat_parallel(mat, method = "emp", normalize = FALSE)
15 | }
16 | \arguments{
17 | \item{mat}{Any discrete numerical matrix, processed column-wise}
18 |
19 | \item{disc}{Name of discretization method. Options include "equalfreq",
20 | "equalwidth", and "globalequalwidth". See \code{infotheo::discretize} for more
21 | detail}
22 |
23 | \item{nbins}{The number of bins by which the continuous data should be
24 | discretized; by default the cube root of the number of samples}
25 |
26 | \item{x, y}{Any discrete numerical vector}
27 |
28 | \item{method}{Name of entropy estimator. Options include "emp", "mm",
29 | "shrink", and "sg". See \code{infotheo::mutinformation} for more detail}
30 |
31 | \item{normalize}{Should the entropies be normalized to fall within the range
32 | [0, 1]?}
33 | }
34 | \value{
35 | Mutual information calculations for filtered matrices
36 | }
37 | \description{
38 | Information Theory
39 | }
40 | \examples{
41 | {
42 | lst_scrna <- CytoTalk::scrna_cyto
43 | pcg <- CytoTalk::pcg_human
44 | cell_type_a <- "Macrophages"
45 | cutoff_a <- 0.8
46 | mat_a <- extract_group(cell_type_a, lst_scrna)
47 | mat_filt_a <- subset_rownames(subset_non_zero(mat_a, cutoff_a), pcg)
48 | mat_disc_a <- discretize_sparse(Matrix::t(mat_filt_a))
49 | result <- mi_mat_parallel(mat_disc_a, method = "mm")
50 | }
51 |
52 | }
53 |
--------------------------------------------------------------------------------
/R/mutinfo.R:
--------------------------------------------------------------------------------
1 | #' @rdname doc_mutinfo
2 | #' @export
3 | discretize_sparse <- function(mat, disc="equalfreq", nbins=nrow(mat)^(1/3)) {
4 | mat_disc <- infotheo::discretize(as.matrix(mat), disc, max(2, nbins))
5 | mat_disc <- t(do.call(rbind, mat_disc)) - 1
6 | Matrix::Matrix(mat_disc, sparse = TRUE)
7 | }
8 |
9 | #' @rdname doc_mutinfo
10 | #' @export
11 | mutinfo_xy <- function(x, y, method="emp", normalize=FALSE) {
12 | if (!normalize) {
13 | return(infotheo::mutinformation(x, y, method))
14 | }
15 |
16 | h1 <- infotheo::entropy(x, method)
17 | h2 <- infotheo::entropy(y, method)
18 | h12 <- infotheo::entropy(data.frame(x, y), method)
19 |
20 | hm <- min(h1, h2)
21 | if (hm == 0) {
22 | return(0)
23 | }
24 |
25 | ((h1 + h2) - h12) / hm
26 | }
27 |
28 | #' @rdname doc_mutinfo
29 | #' @export
30 | mi_mat_parallel <- function(mat, method="emp", normalize=FALSE) {
31 | mat <- as.matrix(mat)
32 | n <- ncol(mat)
33 | ent <- apply(mat, 2, infotheo::entropy, method)
34 |
35 | i <- NULL
36 | res <- foreach::`%dopar%`(foreach::foreach(i = seq_len(n)), {
37 | h1 <- ent[i]
38 | x <- as.numeric(mat[, i])
39 | vapply(i:n, function(j) {
40 | h2 <- ent[j]
41 | y <- as.numeric(mat[, j])
42 | hm <- min(h1, h2)
43 | h12 <- infotheo::entropy(data.frame(x, y), method)
44 | if (hm == 0) {
45 | 0
46 | } else if (normalize) {
47 | ((h1 + h2) - h12) / hm
48 | } else {
49 | ((h1 + h2) - h12)
50 | }
51 | }, numeric(1))
52 | })
53 |
54 | out <- matrix(0, n, n)
55 | out[lower.tri(out, diag = TRUE)] <- unlist(res)
56 | out + t(out * lower.tri(out))
57 | }
58 |
--------------------------------------------------------------------------------
/man/analyze_pathway.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/analysis.R
3 | \name{analyze_pathway}
4 | \alias{analyze_pathway}
5 | \title{Final Network Pathway Analysis}
6 | \usage{
7 | analyze_pathway(df_net_sub, lst_net, cell_type_a, cell_type_b, beta, ntrial)
8 | }
9 | \arguments{
10 | \item{df_net_sub}{A subset of the final network (pathway); for example, the
11 | output of the \code{extract_pathways} function}
12 |
13 | \item{lst_net}{Integrated network}
14 |
15 | \item{cell_type_a}{Name of cell type A that matches scRNA-seq file; for
16 | example, \code{"Fibroblasts"}}
17 |
18 | \item{cell_type_b}{Name of cell type B that matches scRNA-seq file; for
19 | example, \code{"LuminalEpithelialCells"}}
20 |
21 | \item{beta}{Upper limit of the test values of the PCSF objective function
22 | parameter $I^2$, which is inversely proportional to the total number of
23 | genes in a given cell-type pair; suggested to be 100 (default) if the
24 | total number of genes in a given cell-type pair is above 10,000; if the
25 | total number of genes is below 5,000, increase to 500}
26 |
27 | \item{ntrial}{How many empirical simulations to run? (Sample used to form
28 | theoretical Gamma distribution)}
29 | }
30 | \value{
31 | A data-frame containing information relating to pathway size, mean
32 | node prize, mean edge cost, potential scores, and p-values from a fitted
33 | Gamma distribution
34 | }
35 | \description{
36 | Final Network Pathway Analysis
37 | }
38 | \examples{
39 | {
40 | df_net_sub <- result_cyto$pathways$raw[[1]]
41 | lst_net <- result_cyto$integrated_net
42 | cell_type_a <- "Macrophages"
43 | cell_type_b <- "LuminalEpithelialCells"
44 | beta <- 20
45 | ntrial <- 1000
46 | result <- analyze_pathway(df_net_sub, lst_net, cell_type_a, cell_type_b, beta, ntrial)
47 | }
48 |
49 | }
50 |
--------------------------------------------------------------------------------
/DESCRIPTION:
--------------------------------------------------------------------------------
1 | Package: CytoTalk
2 | Title: Signal Transduction Networks from scRNA-seq
3 | Version: 0.99.9
4 | Depends: R (>= 4.1.0)
5 | Authors@R: c(
6 | person("Yuxuan", "Hu",
7 | role = c("aut"),
8 | comment = c(ORCID = "0000-0002-8830-6893")),
9 | person("Shane", "Drabing",
10 | role = c("aut"),
11 | comment = c(ORCID = "0000-0001-9521-6610")),
12 | person("Kai", "Tai",
13 | role = c("cre"),
14 | email = "tanlab4generegulation@gmail.com",
15 | comment = c(ORCID = "0000-0002-9104-5567")))
16 | Description: Perform de novo construction of signal transduction networks using
17 | single-cell transcriptomic data (scRNA-seq). The CytoTalk process
18 | constructs an integrated network of intra- and inter-cellular functional
19 | gene interactions, generating useful graphical output for investigation of
20 | cell-cell communication within and between cell types. CytoTalk attempts to
21 | find the optimal subnetwork in the integrated network that includes genes
22 | with high levels of cell-type-specific expression and close connection to
23 | highly active ligand-receptor pairs. It takes into account metrics like as
24 | Preferential Expression Measure (PEM), mutual information between proteins,
25 | gene relevance, and non-self-talk score.
26 | License: MIT + file LICENSE
27 | Encoding: UTF-8
28 | LazyData: true
29 | Roxygen: list(markdown = TRUE)
30 | RoxygenNote: 7.1.2
31 | Imports:
32 | DiagrammeR,
33 | DiagrammeRsvg,
34 | Matrix,
35 | corpcor,
36 | doParallel,
37 | foreach,
38 | infotheo,
39 | parmigene,
40 | reticulate,
41 | SingleCellExperiment,
42 | tibble,
43 | vroom
44 | Suggests:
45 | markdown,
46 | knitr,
47 | rmarkdown,
48 | testthat (>= 3.0.0)
49 | VignetteBuilder: knitr
50 | biocViews: Software, SingleCell, GeneExpression, Network, Pathways
51 | SystemRequirements:
52 | Cytoscape,
53 | Graphviz,
54 | pcst_fast
55 | Config/testthat/edition: 3
56 |
--------------------------------------------------------------------------------
/man/doc_pcst.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/data.R, R/pcst.R
3 | \name{doc_pcst}
4 | \alias{doc_pcst}
5 | \alias{run_pcst}
6 | \alias{summarize_pcst}
7 | \alias{ks_test_pcst}
8 | \alias{extract_network}
9 | \alias{extract_pathways}
10 | \title{Prize-Collecting Steiner Tree and Pathway Extraction}
11 | \usage{
12 | run_pcst(lst_net, beta_max, omega_min, omega_max)
13 |
14 | summarize_pcst(lst_pcst)
15 |
16 | ks_test_pcst(lst_pcst)
17 |
18 | extract_network(lst_net, lst_pcst, mat_pem, beta, omega)
19 |
20 | extract_pathways(df_net, cell_type_a, depth)
21 | }
22 | \arguments{
23 | \item{lst_net}{Integrated network}
24 |
25 | \item{beta_max}{Upper limit of the test values of the PCSF objective
26 | function parameter $I^2$, which is inversely proportional to the total
27 | number of genes in a given cell-type pair; suggested to be 100 (default)
28 | if the total number of genes in a given cell-type pair is above 10,000; if
29 | the total number of genes is below 5,000, increase to 500}
30 |
31 | \item{omega_min}{Start point of omega range; omega represents the edge cost
32 | of the artificial network, but has been found to be less significant than
33 | beta. Recommended minimum of \code{0.5}}
34 |
35 | \item{omega_max}{End point of range between \code{omega_min} and \code{omega_max},
36 | step size of \code{0.1}. Recommended maximum of \code{1.5}}
37 |
38 | \item{lst_pcst}{PCST output}
39 |
40 | \item{mat_pem}{PEM output}
41 |
42 | \item{beta}{A single beta value, see \code{beta_max} paramter for more detail}
43 |
44 | \item{omega}{A single omega value, see \code{omega_min} for more detail}
45 |
46 | \item{df_net}{Final network; for example, the output of the
47 | \code{extract_network} function}
48 |
49 | \item{cell_type_a}{Name of cell type A that matches scRNA-seq file; for
50 | example, \code{"Fibroblasts"}}
51 |
52 | \item{depth}{Starting at each ligand-receptor pair in the resultant network,
53 | how many steps out from that pair should be taken to generate each
54 | neighborhood?}
55 | }
56 | \value{
57 | Scores for the integrated network
58 | }
59 | \description{
60 | Prize-Collecting Steiner Tree and Pathway Extraction
61 | }
62 | \examples{
63 | {
64 | lst_net <- CytoTalk::result_cyto$integrated_net
65 | beta_max <- 100
66 | omega_min <- 0.5
67 | omega_max <- 0.5
68 | # result <- run_pcst(lst_net, beta_max, omega_min, omega_max)
69 | }
70 |
71 | }
72 |
--------------------------------------------------------------------------------
/man/doc_integrated.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/data.R, R/integrated.R
3 | \name{doc_integrated}
4 | \alias{doc_integrated}
5 | \alias{nonselftalk}
6 | \alias{gene_relevance}
7 | \alias{node_prize}
8 | \alias{crosstalk}
9 | \alias{integrate_network}
10 | \title{Integrated Co-Expression Network}
11 | \usage{
12 | nonselftalk(mat_type, lrp)
13 |
14 | gene_relevance(mat_intra, lrp)
15 |
16 | node_prize(mat_pem, cell_type, vec_relev)
17 |
18 | crosstalk(
19 | mat_pem,
20 | cell_type_a,
21 | cell_type_b,
22 | vec_nst_a,
23 | vec_nst_b,
24 | mat_type,
25 | lrp
26 | )
27 |
28 | integrate_network(
29 | vec_nst_a,
30 | vec_nst_b,
31 | mat_intra_a,
32 | mat_intra_b,
33 | cell_type_a,
34 | cell_type_b,
35 | mat_pem,
36 | mat_type,
37 | lrp
38 | )
39 | }
40 | \arguments{
41 | \item{mat_type}{A matrix of a single cell type}
42 |
43 | \item{lrp}{A dataframe or matrix object with two columns, ligands names and
44 | the names of their receptors; by default, uses the \code{lrp_human} data. This
45 | package also includes \code{lrp_mouse}, but you can also use your own data}
46 |
47 | \item{mat_intra}{Intracellular network of any type}
48 |
49 | \item{mat_pem}{PEM output}
50 |
51 | \item{cell_type}{Name of any cell type}
52 |
53 | \item{vec_relev}{Vector containing gene relevance scores}
54 |
55 | \item{cell_type_a}{Name of cell type A that matches scRNA-seq file; for
56 | example, \code{"Fibroblasts"}}
57 |
58 | \item{cell_type_b}{Name of cell type B that matches scRNA-seq file; for
59 | example, \code{"LuminalEpithelialCells"}}
60 |
61 | \item{vec_nst_a}{Vector containing nonselftalk-scores for cell type A}
62 |
63 | \item{vec_nst_b}{Vector containing nonselftalk-scores for cell type B}
64 |
65 | \item{mat_intra_a}{Intracellular network for cell type A; for example, using
66 | \code{parmigene::aracne.m} on the result of \code{mi_mat_parallel} will filter out
67 | any indirect edges per the data processing inequality}
68 |
69 | \item{mat_intra_b}{Intracellular network for cell type B}
70 | }
71 | \value{
72 | Various outputs to build the integrated network
73 | }
74 | \description{
75 | Integrated Co-Expression Network
76 | }
77 | \examples{
78 | {
79 | lst_scrna <- CytoTalk::scrna_cyto
80 | cell_type_a <- "Macrophages"
81 | lrp <- CytoTalk::lrp_human
82 | mat_a <- extract_group(cell_type_a, lst_scrna)
83 | result <- nonselftalk(mat_a, lrp)
84 | }
85 |
86 | }
87 |
--------------------------------------------------------------------------------
/man/run_cytotalk.Rd:
--------------------------------------------------------------------------------
1 | % Generated by roxygen2: do not edit by hand
2 | % Please edit documentation in R/cytotalk.R
3 | \name{run_cytotalk}
4 | \alias{run_cytotalk}
5 | \title{Main CytoTalk Pipeline}
6 | \usage{
7 | run_cytotalk(
8 | lst_scrna,
9 | cell_type_a,
10 | cell_type_b,
11 | cutoff_a = 0.2,
12 | cutoff_b = 0.2,
13 | pcg = CytoTalk::pcg_human,
14 | lrp = CytoTalk::lrp_human,
15 | beta_max = 100,
16 | omega_min = 0.5,
17 | omega_max = 0.5,
18 | depth = 3,
19 | ntrial = 1000,
20 | cores = NULL,
21 | echo = TRUE,
22 | dir_out = NULL
23 | )
24 | }
25 | \arguments{
26 | \item{lst_scrna}{List containing scRNA-seq data; for example, lists returned
27 | from \code{read_matrix_folder} or \code{read_matrix_with_meta}}
28 |
29 | \item{cell_type_a}{Name of cell type A that matches scRNA-seq file; for
30 | example, \code{"Fibroblasts"}}
31 |
32 | \item{cell_type_b}{Name of cell type B that matches scRNA-seq file; for
33 | example, \code{"LuminalEpithelialCells"}}
34 |
35 | \item{cutoff_a}{Proportional threshold for lowly expressed genes in cell
36 | type A (range of [0-1]); for example, 0.2 means genes with some
37 | expression in at least 20\% of cells are retained}
38 |
39 | \item{cutoff_b}{Proportional expression threshold for cell type B (range of
40 | [0-1])}
41 |
42 | \item{pcg}{A character vector, contains the names of protein coding genes;
43 | by default, uses the \code{pcg_human} data. This package also includes
44 | \code{pcg_mouse}, but you can also use your own data}
45 |
46 | \item{lrp}{A dataframe or matrix object with two columns, ligands names and
47 | the names of their receptors; by default, uses the \code{lrp_human} data. This
48 | package also includes \code{lrp_mouse}, but you can also use your own data}
49 |
50 | \item{beta_max}{Upper limit of the test values of the PCSF objective
51 | function parameter $I^2$, which is inversely proportional to the total
52 | number of genes in a given cell-type pair; suggested to be 100 (default)
53 | if the total number of genes in a given cell-type pair is above 10,000; if
54 | the total number of genes is below 5,000, increase to 500}
55 |
56 | \item{omega_min}{Start point of omega range; omega represents the edge cost
57 | of the artificial network, but has been found to be less significant than
58 | beta. Recommended minimum of \code{0.5}}
59 |
60 | \item{omega_max}{End point of range between \code{omega_min} and \code{omega_max},
61 | step size of \code{0.1}. Recommended maximum of \code{1.5}}
62 |
63 | \item{depth}{Starting at each ligand-receptor pair in the resultant network,
64 | how many steps out from that pair should be taken to generate each
65 | neighborhood?}
66 |
67 | \item{ntrial}{How many random network subsets shall be created to get an
68 | empirical p-value for node prize and edge cost?}
69 |
70 | \item{cores}{How many cores to use for parallel processing?}
71 |
72 | \item{echo}{Should update messages be printed?}
73 |
74 | \item{dir_out}{Folder used for output; if not specified, a "CytoTalk-output"
75 | folder will be generated}
76 | }
77 | \value{
78 | A list containing model parameters, prefential expression measure,
79 | the integrated co-expression network, the results of the PCST, and resulting
80 | pathways from the final extracted network
81 | }
82 | \description{
83 | Main CytoTalk Pipeline
84 | }
85 | \examples{
86 | {
87 | cell_type_a <- "Macrophages"
88 | cell_type_b <- "LuminalEpithelialCells"
89 | cutoff_a <- 0.6
90 | cutoff_b <- 0.6
91 | # result <- CytoTalk::run_cytotalk(CytoTalk::scrna_cyto,
92 | # cell_type_a, cell_type_b,
93 | # cutoff_a, cutoff_b,
94 | # cores = 2)
95 | }
96 |
97 | }
98 |
--------------------------------------------------------------------------------
/R/fileio.R:
--------------------------------------------------------------------------------
1 | #' @noRd
2 | new_named_list <- function(mat, cell_types) {
3 | list(mat = mat, cell_types = cell_types)
4 | }
5 |
6 | #' @noRd
7 | vroom_silent <- function(...) {
8 | suppressMessages(vroom::vroom(..., progress = FALSE))
9 | }
10 |
11 | #' @noRd
12 | vroom_write_silent <- function(x, file, rownames=FALSE) {
13 | x <- as.data.frame(x)
14 | if (rownames) { x <- tibble::rownames_to_column(x) }
15 | suppressMessages(vroom::vroom_write(x, file, progress = FALSE))
16 | }
17 |
18 | #' @noRd
19 | vroom_with_rownames <- function(..., row_names=1) {
20 | dat <- vroom_silent(...)
21 | tibble::column_to_rownames(dat, names(dat)[row_names])
22 | }
23 |
24 | #' @noRd
25 | vroom_sparse_with_rownames <- function(..., row_names=1) {
26 | dat <- vroom_with_rownames(..., row_names = row_names)
27 | Matrix::Matrix(Matrix::as.matrix(dat), sparse = TRUE)
28 | }
29 |
30 | #' Convert SingleCellExperiment to Named List
31 | #'
32 | #' @param sce SingleCellExperiment object
33 | #'
34 | #' @return A named list containing a sparse data matrix and cell type metadata
35 | #'
36 | #' @export
37 | from_single_cell_experiment <- function(sce) {
38 | count <- SingleCellExperiment::logcounts(sce)
39 | names <- colnames(sce)
40 | new_named_list(count, names)
41 | }
42 |
43 | #' Read Folder with scRNAseq Data
44 | #'
45 | #' @param dpath The path of a directory, which contains scRNAseq matrices
46 | #'
47 | #' @param pattern A regular expression, matches scRNAseq filenames
48 | #'
49 | #' @param auto_transform Should count data be transformed if detected?
50 | #'
51 | #' @examples {
52 | #' dir_in <- "~/Tan-Lab/scRNAseq-data"
53 | #' # lst_scrna <- CytoTalk::read_matrix_folder(dir_in)
54 | #' # result <- str(lst_scrna$cell_types)
55 | #' }
56 | #'
57 | #' @return A named list containing a sparse data matrix and cell type metadata
58 | #'
59 | #' @export
60 | read_matrix_folder <- function(
61 | dpath, pattern=".*scRNAseq_(.+?)\\..+", auto_transform=TRUE) {
62 |
63 | # initial parameters
64 | mat <- NULL
65 | msg <- "not all rownames identical between input files"
66 |
67 | # determine filepaths
68 | fpaths <- dir_full(dpath, pattern = pattern)
69 |
70 | # read in all files
71 | for (fpath in fpaths) {
72 | cell_type <- gsub(pattern, "\\1", fpath)
73 | if (is.null(mat)) {
74 | # read in
75 | mat <- vroom_sparse_with_rownames(fpath)
76 | # start cell types vector
77 | cell_types <- rep(cell_type, ncol(mat))
78 | } else {
79 | # read in
80 | new <- vroom_sparse_with_rownames(fpath)
81 | # check for identical rownames
82 | errorifnot(identical(rownames(mat), rownames(new)), msg)
83 | # accumulate cell type names
84 | cell_types <- c(cell_types, rep(cell_type, ncol(new)))
85 | # combine all data
86 | mat <- cbind(mat, new)
87 | }
88 | }
89 |
90 | # check for count data
91 | mat <- check_count_data(mat, auto_transform)
92 |
93 | # return sparse matrix
94 | new_named_list(mat, cell_types)
95 | }
96 |
97 | #' Read scRNAseq Data Matrix and Metadata
98 | #'
99 | #' @param fpath_mat The path of a file containing a scRNAseq data matrix
100 | #'
101 | #' @param fpath_meta The path of a file contianing column metadata (cell types)
102 | #'
103 | #' @param auto_transform Should count data be transformed if detected?
104 | #'
105 | #' @examples {
106 | #' fpath_mat <- "~/Tan-Lab/scRNAseq-data-cpdb/sample_counts.txt"
107 | #' fpath_meta <- "~/Tan-Lab/scRNAseq-data-cpdb/sample_meta.txt"
108 | #' # lst_scrna <- CytoTalk::read_matrix_with_meta(fpath_mat, fpath_meta)
109 | #' # result <- str(lst_scrna$cell_types)
110 | #' }
111 | #'
112 | #' @return A named list containing a sparse data matrix and cell type metadata
113 | #'
114 | #' @export
115 | read_matrix_with_meta <- function(fpath_mat, fpath_meta, auto_transform=TRUE) {
116 | # read in
117 | mat <- vroom_sparse_with_rownames(fpath_mat)
118 | meta <- vroom_with_rownames(fpath_meta)
119 |
120 | # match meta to matrix
121 | index <- match(rownames(meta), colnames(mat))
122 | # ensure index matches exactly
123 | errorifnot(!any(is.na(index)), "meta file does not match matrix colnames")
124 | # otherwise, reorder
125 | cell_types <- meta[index, 1]
126 |
127 | # check for count data
128 | mat <- check_count_data(mat, auto_transform)
129 |
130 | # return sparse matrix
131 | new_named_list(mat, cell_types)
132 | }
133 |
--------------------------------------------------------------------------------
/R/analysis.R:
--------------------------------------------------------------------------------
1 | #' @noRd
2 | score_subnetwork <- function(prizes, costs, beta) {
3 | potential <- (beta * sum(prizes) - sum(costs))
4 | c(mean(prizes), mean(costs), potential)
5 | }
6 |
7 | #' @noRd
8 | subsample_network_shuffle <- function(prizes, costs, n_nodes, n_edges) {
9 | list(prize = sample(prizes, n_nodes), cost = sample(costs, n_edges))
10 | }
11 |
12 | #' @noRd
13 | gamma_fit <- function(x, y) {
14 | x <- x - min(x) + y
15 | shape <- mean(x)^2 / stats::var(x)
16 | scale <- stats::var(x) / mean(x)
17 | list(
18 | params = c(shape, scale),
19 | kstest = stats::ks.test(x, "pgamma", shape = shape, scale = scale)
20 | )
21 | }
22 |
23 | #' @noRd
24 | gamma_score <- function(x, inverse=FALSE) {
25 | if (inverse) { x <- (-x) }
26 |
27 | v <- x[-1]
28 | y <- stats::optimize(function(y) {
29 | -gamma_fit(v, y)$kstest$p.value
30 | }, c(0, 1000))$minimum
31 |
32 | fit <- gamma_fit(v, y)
33 | shape <- fit$params[1]
34 | scale <- fit$params[2]
35 |
36 | x1 <- x[1] - min(v) + y
37 | stats::pgamma(x1, shape = shape, scale = scale, lower.tail = FALSE)
38 | }
39 |
40 | #' @noRd
41 | convert_names <- function(x, cell_type_a, cell_type_b) {
42 | suffix_a <- sprintf("__%s", cell_type_a)
43 | suffix_b <- sprintf("__%s", cell_type_b)
44 | gsub(suffix_a, "", gsub(suffix_b, "_", x))
45 | }
46 |
47 | #' Final Network Pathway Analysis
48 | #'
49 | #' @param df_net_sub A subset of the final network (pathway); for example, the
50 | #' output of the `extract_pathways` function
51 | #'
52 | #' @param lst_net Integrated network
53 | #'
54 | #' @param cell_type_a Name of cell type A that matches scRNA-seq file; for
55 | #' example, `"Fibroblasts"`
56 | #'
57 | #' @param cell_type_b Name of cell type B that matches scRNA-seq file; for
58 | #' example, `"LuminalEpithelialCells"`
59 | #'
60 | #' @param beta Upper limit of the test values of the PCSF objective function
61 | #' parameter $I^2$, which is inversely proportional to the total number of
62 | #' genes in a given cell-type pair; suggested to be 100 (default) if the
63 | #' total number of genes in a given cell-type pair is above 10,000; if the
64 | #' total number of genes is below 5,000, increase to 500
65 | #'
66 | #' @param ntrial How many empirical simulations to run? (Sample used to form
67 | #' theoretical Gamma distribution)
68 | #'
69 | #' @examples {
70 | #' df_net_sub <- result_cyto$pathways$raw[[1]]
71 | #' lst_net <- result_cyto$integrated_net
72 | #' cell_type_a <- "Macrophages"
73 | #' cell_type_b <- "LuminalEpithelialCells"
74 | #' beta <- 20
75 | #' ntrial <- 1000
76 | #' result <- analyze_pathway(df_net_sub, lst_net, cell_type_a, cell_type_b, beta, ntrial)
77 | #' }
78 | #'
79 | #' @return A data-frame containing information relating to pathway size, mean
80 | #' node prize, mean edge cost, potential scores, and p-values from a fitted
81 | #' Gamma distribution
82 | #'
83 | #' @export
84 | analyze_pathway <- function(
85 | df_net_sub, lst_net, cell_type_a, cell_type_b, beta, ntrial) {
86 |
87 | # extract node and tables
88 | df_net_nodes <- lst_net$nodes[, c(1, 2)]
89 | df_net_edges <- lst_net$edges[, c(1, 2, 3)]
90 |
91 | # set columns numeric
92 | df_net_nodes$prize <- as.numeric(df_net_nodes$prize)
93 | df_net_edges$cost <- as.numeric(df_net_edges$cost)
94 |
95 | # convert names
96 | df_net_nodes$node <- convert_names(
97 | df_net_nodes$node, cell_type_a, cell_type_b)
98 | df_net_edges$node1 <- convert_names(
99 | df_net_edges$node1, cell_type_a, cell_type_b)
100 | df_net_edges$node2 <- convert_names(
101 | df_net_edges$node2, cell_type_a, cell_type_b)
102 |
103 | # prepare nodes
104 | df_node <- data.frame(
105 | node = c(df_net_sub$node1, df_net_sub$node2),
106 | prize = c(df_net_sub$node1_prize, df_net_sub$node2_prize)
107 | )
108 | df_node <- df_node[!duplicated(df_node[, 1]), ]
109 | n_nodes <- nrow(df_node)
110 |
111 | # prepare edges
112 | df_edge <- df_net_sub[, c("node1", "node2", "cost")]
113 | n_edges <- nrow(df_edge)
114 |
115 | # begin scores
116 | scores <- score_subnetwork(df_node[, 2], df_edge[, 3], beta)
117 |
118 | # simulate random subsets
119 | for (i in seq_len(ntrial)) {
120 | lst <- subsample_network_shuffle(
121 | df_net_nodes[, 2], df_net_edges[, 3], n_nodes, n_edges)
122 | scores <- rbind(scores, score_subnetwork(lst[[1]], lst[[2]], beta))
123 | }
124 |
125 | # new row of data
126 | data.frame(
127 | num_edges = n_edges, num_nodes = nrow(df_node),
128 | mean_prize = scores[1, 1], mean_cost = scores[1, 2],
129 | potential = scores[1, 3],
130 | pval_prize = gamma_score(scores[, 1]),
131 | pval_cost = gamma_score(scores[, 2], inverse = TRUE),
132 | pval_potential = gamma_score(scores[, 3])
133 | )
134 | }
135 |
--------------------------------------------------------------------------------
/R/graphing.R:
--------------------------------------------------------------------------------
1 | # CONSTANTS
2 |
3 |
4 | HEX <- c(0:9, LETTERS[seq_len(6)])
5 | URL_GENECARDS <- "https://www.genecards.org/cgi-bin/carddisp.pl?gene="
6 | URL_WIKIPI <- "https://hagrid.dbmi.pitt.edu/wiki-pi/index.php/search?q="
7 | FORM_NODE <- paste0(
8 | "\"%s\" [label = \"%s\" href = \"",
9 | URL_GENECARDS,
10 | "%s\" width = %s height = %s fillcolor = \"%s\"]"
11 | )
12 | FORM_EDGE <- paste0(
13 | "\"%s\" -> \"%s\" [href=\"",
14 | URL_WIKIPI,
15 | "%s+%s\" penwidth=%s style=%s]"
16 | )
17 | FORM_GV <- trimws("
18 | digraph {\n
19 | pad=0.25
20 | layout=dot
21 | labeljust=l
22 | splines=true
23 | rankdir=TB
24 | ranksep=1
25 | nodesep=0.1
26 | compound=true
27 | outputorder=\"edgesfirst\"\n
28 | node [shape=oval fixedsize=true target=\"_blank\"]
29 | node [fontname=\"Arial\" fontsize=9 style=filled ordering=out]
30 | edge [arrowhead=none target=\"_blank\"]\n
31 | subgraph cluster0 {\n
32 | margin=20
33 | color=none
34 | style=filled
35 | fillcolor=\"#EEEEEE\"\n
36 | %s\n
37 | }\n
38 | subgraph cluster1 {\n
39 | margin=20
40 | color=none
41 | style=filled
42 | fillcolor=\"#EEEEEE\"\n
43 | %s\n
44 | }\n
45 | // cluster external horizontal order
46 | %s
47 | // cluster external
48 | edge [color=limegreen arrowhead=normal arrowtail=normal dir=back]
49 | %s\n
50 | }")
51 |
52 |
53 | # FUNCTIONS
54 |
55 |
56 | #' @rdname doc_graphing
57 | #' @export
58 | write_network_sif <- function(df_net, cell_type_a, dir_out) {
59 | # format dir path
60 | dir_out_cs <- file.path(dir_out, "cytoscape")
61 |
62 | # make sure it exists
63 | if (!dir.exists(dir_out_cs)) {
64 | dir.create(dir_out_cs, recursive = TRUE)
65 | }
66 |
67 | # format filepaths
68 | fpath_node <- file.path(dir_out_cs, "CytoscapeNodes.txt")
69 | fpath_edge <- file.path(dir_out_cs, "CytoscapeEdges.txt")
70 | fpath_sif <- file.path(dir_out_cs, "CytoscapeNetwork.sif")
71 |
72 | # make names unique
73 | df_net$node1 <- paste0(
74 | df_net$node1, ifelse(df_net$node1_type == cell_type_a, "", "_"))
75 | df_net$node2 <- paste0(
76 | df_net$node2, ifelse(df_net$node2_type == cell_type_a, "", "_"))
77 |
78 | # format edges
79 | edge_types <- ifelse(df_net$is_ct_edge, "pr", "pp")
80 | edge_names <- sprintf("%s (%s) %s", df_net$node1, edge_types, df_net$node2)
81 | edges <- gsub("[()]", "", edge_names)
82 |
83 | # create node table
84 | index1 <- c("node1", "node1_type", "node1_prize", "node1_pem")
85 | index2 <- c("node2", "node2_type", "node2_prize", "node2_pem")
86 | df_node <- data.frame(rbind(
87 | as.matrix(df_net[, index1]),
88 | as.matrix(df_net[, index2])
89 | ))
90 |
91 | # naming and type conversion
92 | names(df_node) <- c("node", "type", "prize", "pem")
93 | df_node <- utils::type.convert(df_node, as.is = TRUE)
94 |
95 | # create edge table
96 | df_edge <- cbind(edge_names, df_net[, c("cost", "is_ct_edge")])
97 | names(df_edge) <- c("edge", "cost", "is_ct_edge")
98 |
99 | # write out sif
100 | writeLines(paste(edges, collapse = "\n"), fpath_sif)
101 |
102 | # write out node and edge tables
103 | vroom_write_silent(df_node, fpath_node)
104 | vroom_write_silent(df_edge, fpath_edge)
105 | NULL
106 | }
107 |
108 | #' @rdname doc_graphing
109 | #' @export
110 | graph_pathway <- function(df_net_sub) {
111 | # reorder nodes
112 | df_cpy <- df_net_sub
113 | index_lt <- (df_net_sub$node1_pem < df_net_sub$node2_pem)
114 | index1 <- c("node1", "node1_type", "node1_prize", "node1_pem")
115 | index2 <- c("node2", "node2_type", "node2_prize", "node2_pem")
116 | for (i in seq_len(length(index_lt))) {
117 | if (index_lt[i]) {
118 | df_net_sub[i, index2] <- df_cpy[i, index1]
119 | df_net_sub[i, index1] <- df_cpy[i, index2]
120 | }
121 | }
122 |
123 | # reorder edges
124 | df_net_sub <- df_net_sub[order(as.numeric(df_net_sub$cost)), ]
125 |
126 | # normalize PEM
127 | index <- c("node1_pem", "node2_pem")
128 | df_net_sub[index] <- apply(df_net_sub[index], 2, function(x) {
129 | minmax(ifelse(x < 0 | is.na(x), min(x, na.rm = TRUE), x))
130 | })
131 |
132 | # prepare nodes
133 | df_node <- data.frame(rbind(
134 | as.matrix(df_net_sub[, index1]),
135 | as.matrix(df_net_sub[, index2])
136 | ))
137 |
138 | df_node <- df_node[!duplicated(df_node), ]
139 | df_node <- utils::type.convert(df_node, as.is = TRUE)
140 | names(df_node) <- c("node", "type", "prize", "pem")
141 |
142 | # string format nodes
143 | ew <- function(x) endsWith(x, "_")
144 | index_nodes <- ew(df_node$node)
145 | clean <- trimws(df_node$node, whitespace = "_")
146 | size <- 0.5 + 3.5 * df_node$prize^2.5
147 | color <- grDevices::hsv(
148 | ifelse(ew(df_node$node), 0.02, 0.55), (df_node$pem), 1
149 | )
150 | nodes <- sprintf(
151 | FORM_NODE, df_node$node, clean, clean, size, size, color)
152 |
153 | # string format edges
154 | index_edges <- ew(df_net_sub$node1) + ew(df_net_sub$node2)
155 | c1 <- trimws(df_net_sub$node1, whitespace = "_")
156 | c2 <- trimws(df_net_sub$node2, whitespace = "_")
157 | size <- 1.25 + 3.75 * (1 - df_net_sub$cost)^2
158 | # size <- ifelse(df_net_sub$is_ct_edge, 2, 1) * size
159 | style <- ifelse(df_net_sub$is_ct_edge, "dashed", "solid")
160 | edges <- sprintf(
161 | FORM_EDGE, df_net_sub$node1, df_net_sub$node2, c1, c2, size, style
162 | )
163 |
164 | # cell type names
165 | type_a <- df_node[!ew(df_node$node), "type"][1]
166 | type_b <- df_node[ew(df_node$node), "type"][1]
167 |
168 | # string format graph
169 | graph <- sprintf(FORM_GV,
170 | paste0(
171 | sprintf("label=\"%s\"\ntooltip=\"%s\"\n", type_a, type_a),
172 | paste0(nodes[!index_nodes], collapse = "\n"), "\n",
173 | paste0(edges[index_edges == 0], collapse = "\n"),
174 | collapse = "\n"
175 | ),
176 | paste0(
177 | sprintf("label=\"%s\"\ntooltip=\"%s\"\n", type_b, type_b),
178 | paste0(nodes[index_nodes], collapse = "\n"), "\n",
179 | paste0(edges[index_edges == 2], collapse = "\n"),
180 | collapse = "\n"
181 | ),
182 | sprintf(
183 | "%s [style=invis, constraint=true]\n",
184 | gsub("\\s+\\[.+", "", edges[index_edges == 1][1])
185 | ),
186 | paste0(
187 | edges[index_edges == 1],
188 | collapse = "\n"
189 | )
190 | )
191 |
192 | # return out
193 | DiagrammeR::grViz(graph)
194 | }
195 |
--------------------------------------------------------------------------------
/R/matrix.R:
--------------------------------------------------------------------------------
1 | #' @noRd
2 | is_integer <- function(x) {
3 | identical(x, round(x))
4 | }
5 |
6 | #' @noRd
7 | zero_diag <- function(mat) {
8 | diag(mat) <- 0
9 | mat
10 | }
11 |
12 | #' @noRd
13 | img <- function(mat, ..., scl=0) {
14 | mat <- as.matrix(mat)
15 | diag(mat) <- diag(mat) * scl
16 | graphics::image(mat, ...)
17 | }
18 |
19 | #' @noRd
20 | proportion_non_zero <- function(mat) {
21 | row_props <- Matrix::rowSums(mat != 0) / ncol(mat)
22 | return(row_props)
23 | }
24 |
25 | #' Subset Rows on Proportion Non-Zero
26 | #'
27 | #' @param mat A numerical matrix
28 | #'
29 | #' @param cutoff Threshold value in range \[0, 1\]; proportion of each row
30 | #' required to be non-zero
31 | #'
32 | #' @examples {
33 | #' lst_scrna <- CytoTalk::scrna_cyto
34 | #' cell_type_a <- "Macrophages"
35 | #' cutoff_a <- 0.8
36 | #' mat_a <- extract_group(cell_type_a, lst_scrna)
37 | #' result <- subset_non_zero(mat_a, cutoff_a)
38 | #' }
39 | #'
40 | #' @return A matrix
41 | #'
42 | #' @export
43 | subset_non_zero <- function(mat, cutoff) {
44 | index <- (cutoff <= proportion_non_zero(mat))
45 | mat[index, ]
46 | }
47 |
48 | #' @noRd
49 | subset_non_zero_old <- function(mat, cutoff) {
50 | thresh <- floor(ncol(mat) * cutoff)
51 | index <- thresh <= Matrix::rowSums(mat != 0)
52 | mat[index, ]
53 | }
54 |
55 | #' Subset Rows on Rownames
56 | #'
57 | #' @param mat A numerical matrix
58 | #'
59 | #' @param labels A subset of the matrix's rownames to filter by
60 | #'
61 | #' @examples {
62 | #' lst_scrna <- CytoTalk::scrna_cyto
63 | #' pcg <- CytoTalk::pcg_human
64 | #' cell_type_a <- "Macrophages"
65 | #' mat_a <- extract_group(cell_type_a, lst_scrna)
66 | #' result <- subset_rownames(mat_a, pcg)
67 | #' }
68 | #'
69 | #' @return A matrix
70 | #'
71 | #' @export
72 | subset_rownames <- function(mat, labels) {
73 | index <- which(!is.na(cmatch(rownames(mat), labels)))
74 | mat[index, ]
75 | }
76 |
77 | #' @noRd
78 | add_noise <- function(mat) {
79 | n <- nrow(mat)
80 | m <- ncol(mat)
81 | dummy <- c(1e-20, rep(0, m - 1))
82 | mat <- mat + matrix(replicate(n, sample(dummy)), n, byrow = TRUE)
83 | Matrix::Matrix(mat)
84 | }
85 |
86 | #' @noRd
87 | extract_group_basic <- function(group, mat, labels) {
88 | mat[, labels == group, drop = FALSE]
89 | }
90 |
91 | #' Extract Cell Type from Meta List
92 | #'
93 | #' @param group Character vector with a single string, name of cell type to
94 | #' extract
95 | #'
96 | #' @param lst A meta matrix list (outputted from `doc_fileio` methods)
97 | #'
98 | #' @examples {
99 | #' lst_scrna <- CytoTalk::scrna_cyto
100 | #' cell_type_a <- "Macrophages"
101 | #' result <- extract_group(cell_type_a, lst_scrna)
102 | #' }
103 | #'
104 | #' @return A matrix
105 | #'
106 | #' @export
107 | extract_group <- function(group, lst) {
108 | extract_group_basic(group, lst[[1]], lst[[2]])
109 | }
110 |
111 | #' @noRd
112 | group_meta_basic <- function(mat, labels) {
113 | groups <- sort(unique(labels))
114 | lst <- lapply(groups, extract_group_basic, mat, labels)
115 | names(lst) <- groups
116 | lst
117 | }
118 |
119 | #' Meta List to Named List
120 | #'
121 | #' @param lst A meta matrix list (outputted from `doc_fileio` methods)
122 | #'
123 | #' @examples {
124 | #' lst_scrna <- CytoTalk::scrna_cyto
125 | #' result <- group_meta(lst_scrna)
126 | #' }
127 | #'
128 | #' @return A list of matrices
129 | #'
130 | #' @export
131 | group_meta <- function(lst) {
132 | group_meta_basic(lst[[1]], lst[[2]])
133 | }
134 |
135 | #' Named List to Meta List
136 | #'
137 | #' @param lst A meta matrix list (outputted from `doc_fileio` methods)
138 | #'
139 | #' @examples {
140 | #' lst_scrna <- CytoTalk::scrna_cyto
141 | #' lst_group <- group_meta(lst_scrna)
142 | #' result <- ungroup_meta(lst_group)
143 | #' }
144 | #'
145 | #' @return A list of a matrix and a meta vector
146 | #'
147 | #' @export
148 | ungroup_meta <- function(lst) {
149 | ncols <- vapply(lst, ncol, numeric(1))
150 | cell_types <- unlist(lapply(seq_len(length(ncols)), function(i) {
151 | rep(names(ncols)[i], ncols[i])
152 | }))
153 | new_named_list(do.call(cbind, lst), cell_types)
154 | }
155 |
156 | #' Find Ligand Receptor Pairs in Matrix
157 | #'
158 | #' @param mat A numerical matrix
159 | #'
160 | #' @param lrp Ligand-receptor pair data.frame; see `CytoTalk::lrp_human` for
161 | #' an example
162 | #'
163 | #' @examples {
164 | #' lst_scrna <- CytoTalk::scrna_cyto
165 | #' lrp <- CytoTalk::lrp_human
166 | #' cell_type_a <- "Macrophages"
167 | #' mat_a <- extract_group(cell_type_a, lst_scrna)
168 | #' result <- match_lr_pairs(mat_a, lrp)
169 | #' }
170 | #'
171 | #' @return A matrix with two columns (ligand-receptor)
172 | #'
173 | #' @export
174 | match_lr_pairs <- function(mat, lrp) {
175 | # save a copy of the rownames
176 | hold <- rownames(mat)
177 |
178 | # match lr_pairs to mat rownames
179 | index <- data.frame(apply(lrp, 2, function(x) {
180 | match(toupper(x), toupper(hold))
181 | }))
182 | index <- index[rowSums(is.na(index)) == 0, ]
183 |
184 | # return out
185 | data.frame(
186 | ligand = hold[index[, 1]],
187 | receptor = hold[index[, 2]]
188 | )
189 | }
190 |
191 | #' scRNAseq Count Normalization
192 | #'
193 | #' @param mat An integer matrix
194 | #'
195 | #' @param scale.factor A single number constant by which to scale the
196 | #' transformed data by
197 | #'
198 | #' @examples {
199 | #' lst_scrna <- CytoTalk::scrna_cyto
200 | #' cell_type_a <- "Macrophages"
201 | #' mat_a <- extract_group(cell_type_a, lst_scrna)
202 | #' result <- normalize_sparse(mat_a)
203 | #' }
204 | #'
205 | #' @return A matrix
206 | #'
207 | #' @export
208 | normalize_sparse <- function(mat, scale.factor=10000) {
209 | log1p(Matrix::t(Matrix::t(mat) / Matrix::colSums(mat) * scale.factor))
210 | }
211 |
212 | #' Transform Count Data if Detected
213 | #'
214 | #' @param mat A numerical matrix
215 | #'
216 | #' @param auto_transform Should the data be transformed if counts are detected?
217 | #'
218 | #' @examples {
219 | #' lst_scrna <- CytoTalk::scrna_cyto
220 | #' cell_type_a <- "Macrophages"
221 | #' mat_a <- extract_group(cell_type_a, lst_scrna)
222 | #' result <- check_count_data(mat_a)
223 | #' }
224 | #'
225 | #' @return A matrix
226 | #'
227 | #' @export
228 | check_count_data <- function(mat, auto_transform=TRUE) {
229 | check <- is_integer(mat)
230 | if (check) {
231 | if (auto_transform) {
232 | mat <- normalize_sparse(mat)
233 | # warn that normalization was performed
234 | msg <- paste(
235 | "count data detected;",
236 | "auto-transformed (see `?check_count_data`)"
237 | )
238 | warnifnot(!check, msg)
239 | } else {
240 | # warn if counts detected
241 | msg <- "count data detected, make sure to transform it"
242 | warnifnot(!check, msg)
243 | }
244 | }
245 | # return matrix
246 | mat
247 | }
248 |
--------------------------------------------------------------------------------
/R/integrated.R:
--------------------------------------------------------------------------------
1 | #' @rdname doc_integrated
2 | #' @export
3 | nonselftalk <- function(mat_type, lrp) {
4 | # make sure function takes in a Matrix type
5 | mat_type <- Matrix::Matrix(mat_type)
6 |
7 | lrp_index <- match_lr_pairs(mat_type, lrp)
8 | errorifnot(0 < nrow(lrp_index), "no ligand-receptor pairs found")
9 |
10 | index <- which(Matrix::rowSums(mat_type != 0) == 0)
11 |
12 | mat_type[index, ] <- add_noise(mat_type[index, ])
13 | mat_disc <- discretize_sparse(
14 | Matrix::t(mat_type), "equalwidth", max(2, ncol(mat_type)^(1/2)))
15 |
16 | mi <- NULL
17 | for (i in seq_len(nrow(lrp_index))) {
18 | row <- unlist(lrp_index[i, ])
19 | x <- mat_disc[, row[1]]
20 | y <- mat_disc[, row[2]]
21 | mi <- c(mi, mutinfo_xy(x, y, "mm", TRUE))
22 | }
23 |
24 | -log10(ifelse(mi < 0, 0, mi))
25 | }
26 |
27 | #' @rdname doc_integrated
28 | #' @export
29 | gene_relevance <- function(mat_intra, lrp) {
30 | # create a new matrix, same size
31 | mat_nsq <- as.matrix(mat_intra) * 0
32 | # set the diagonal as the network rowsums
33 | diag(mat_nsq) <- Matrix::rowSums(mat_intra)
34 |
35 | # negative square root
36 | mat_nsq <- corpcor::mpower(mat_nsq, -0.5)
37 | # symmetric matrix
38 | mat_wnorm <- mat_nsq %*% as.matrix(mat_intra) %*% mat_nsq
39 | # match genes to LR pair
40 | lrp_index <- !is.na(cmatch(rownames(mat_intra), unlist(lrp)))
41 |
42 | # compute gene relevance value,
43 | # random walk with restart
44 | n <- 50
45 | alpha <- 0.9
46 | offset <- (1 - alpha) * lrp_index
47 | mat_relev <- as.matrix(lrp_index)
48 |
49 | for (i in seq_len(n)) {
50 | mat_relev <- alpha * (mat_wnorm %*% mat_relev) + offset
51 | }
52 |
53 | vec_relev <- as.numeric(mat_relev)
54 | names(vec_relev) <- rownames(mat_intra)
55 |
56 | vec_relev
57 | }
58 |
59 | #' @rdname doc_integrated
60 | #' @export
61 | node_prize <- function(mat_pem, cell_type, vec_relev) {
62 | # match PEM cell type
63 | vec_pem <- mat_pem[, cell_type == colnames(mat_pem)]
64 |
65 | # match gene relevance names to cell type vector
66 | index <- match(toupper(names(vec_relev)), toupper(names(vec_pem)))
67 | vec_pem_match <- vec_pem[index]
68 |
69 | # relevance times cell specific,
70 | # if negative then zero out
71 | vec_relev * ifelse(vec_pem_match < 0, 0, vec_pem_match)
72 | }
73 |
74 | #' @rdname doc_integrated
75 | #' @export
76 | crosstalk <- function(
77 | mat_pem, cell_type_a, cell_type_b, vec_nst_a, vec_nst_b, mat_type, lrp) {
78 |
79 | # grab relevant PEM scores, zero out negatives and NaNs
80 | vec_pem_names <- rownames(mat_pem)
81 | vec_pem_a <- zero_na_neg(mat_pem[, cell_type_a])
82 | vec_pem_b <- zero_na_neg(mat_pem[, cell_type_b])
83 |
84 | # zero out bad NST scores, merge the them together
85 | df_nst <- match_lr_pairs(mat_type, lrp)
86 | df_nst[, "mi_a"] <- zero_na_neg(vec_nst_a)
87 | df_nst[, "mi_b"] <- zero_na_neg(vec_nst_b)
88 |
89 | # initialize variables
90 | df_ct <- data.frame()
91 |
92 | for (i in seq_len(nrow(df_nst))) {
93 | # find the LR pair
94 | lig <- df_nst[i, "ligand"]
95 | rec <- df_nst[i, "receptor"]
96 |
97 | # non-self talk score
98 | scr_nst <- sum(df_nst[i, c("mi_a", "mi_b")]) / 2
99 |
100 | # expressed score
101 | scr_expr <- (vec_pem_a[lig] + vec_pem_b[rec]) / 2
102 |
103 | df_ct <- rbind(df_ct, data.frame(
104 | ligand = lig, receptor = rec, ligand_type = cell_type_a,
105 | receptor_type = cell_type_b, nst = scr_nst, expr = scr_expr
106 | ))
107 |
108 | # from type B to A (sometimes skipped)
109 | if (!identical(lig, rec)) {
110 | # expressed score (notice the difference!)
111 | scr_expr <- (vec_pem_b[lig] + vec_pem_a[rec]) / 2
112 |
113 | df_ct <- rbind(df_ct, data.frame(
114 | ligand = lig, receptor = rec, ligand_type = cell_type_b,
115 | receptor_type = cell_type_a, nst = scr_nst, expr = scr_expr
116 | ))
117 | }
118 | }
119 |
120 | # compute crosstalk score
121 | df_ct[, "crosstalk"] <- minmax(df_ct[, "expr"]) * minmax(df_ct[, "nst"])
122 | rownames(df_ct) <- NULL
123 | df_ct
124 | }
125 |
126 | #' @noRd
127 | extract_lower_nonzero <- function(mat) {
128 | index <- Matrix::which(
129 | lower.tri(mat, diag = TRUE) & mat != 0, arr.ind = TRUE
130 | )
131 | df <- data.frame(apply(index, 2, function(x) rownames(mat)[x]))
132 | df[, "val"] <- mat[index]
133 | df
134 | }
135 |
136 | #' @rdname doc_integrated
137 | #' @export
138 | integrate_network <- function(
139 | vec_nst_a, vec_nst_b, mat_intra_a, mat_intra_b,
140 | cell_type_a, cell_type_b, mat_pem, mat_type, lrp) {
141 |
142 | # gene relevance
143 | vec_gr_a <- gene_relevance(mat_intra_a, lrp)
144 | vec_gr_b <- gene_relevance(mat_intra_b, lrp)
145 |
146 | # node prize
147 | vec_np_a <- node_prize(mat_pem, cell_type_a, vec_gr_a)
148 | vec_np_b <- node_prize(mat_pem, cell_type_b, vec_gr_b)
149 |
150 | # crosstalk
151 | df_edge_ct <- crosstalk(
152 | mat_pem, cell_type_a, cell_type_b, vec_nst_a, vec_nst_b, mat_type, lrp)
153 |
154 | # extract MI scores from sparse matrix
155 | df_edge_a <- extract_lower_nonzero(mat_intra_a)
156 | df_edge_b <- extract_lower_nonzero(mat_intra_b)
157 |
158 | # casefold names, determine node and edge types
159 | node_names_a <- add_suffix(names(vec_np_a), cell_type_a)
160 | node_names_b <- add_suffix(names(vec_np_b), cell_type_b)
161 | edge_names_a <- apply(df_edge_a[, c(1, 2)], 2, add_suffix, cell_type_a)
162 | edge_names_b <- apply(df_edge_b[, c(1, 2)], 2, add_suffix, cell_type_b)
163 | edge_names_ct <- cbind(
164 | add_suffix(df_edge_ct[, "ligand"], df_edge_ct[, "ligand_type"]),
165 | add_suffix(df_edge_ct[, "receptor"], df_edge_ct[, "receptor_type"])
166 | )
167 |
168 | # compile full node names
169 | node_names_full <- unique(c(node_names_a, node_names_b))
170 |
171 | # validate crosstalk edges
172 | index_ct <- rowSums(apply(edge_names_ct, 2, "%in%", node_names_full)) == 2
173 |
174 | # compile full edges and costs
175 | edge_full <- rbind(edge_names_a, edge_names_b, edge_names_ct[index_ct, ])
176 | cost <- list(
177 | df_edge_a$val, df_edge_b$val, df_edge_ct[index_ct, "crosstalk"])
178 |
179 | # normalize edge costs
180 | cost_norm <- unlist(lapply(cost, scale))
181 | cost_norm[is.na(cost_norm)] <- 0
182 | cost_full <- 1 - minmax(cost_norm)
183 |
184 | # grab relevant PEM scores,
185 | # zero out negatives and NaNs
186 | vec_pem_names <- rownames(mat_pem)
187 | vec_pem_a <- zero_na_neg(mat_pem[, cell_type_a])
188 | vec_pem_b <- zero_na_neg(mat_pem[, cell_type_b])
189 |
190 | # match PEM
191 | vec_pem_match_a <- vec_pem_a[cmatch(names(vec_np_a), names(vec_pem_a))]
192 | vec_pem_match_b <- vec_pem_b[cmatch(names(vec_np_b), names(vec_pem_b))]
193 |
194 | # node dataframe
195 | df_net_node <- data.frame(
196 | node = c(node_names_a, node_names_b),
197 | prize = c(vec_np_a, vec_np_b),
198 | pem = c(vec_pem_match_a, vec_pem_match_b),
199 | gene_relevance = c(vec_gr_a, vec_gr_b)
200 | )
201 |
202 | # edge dataframe
203 | df_net_edge <- data.frame(
204 | node1 = edge_full[, 1],
205 | node2 = edge_full[, 2],
206 | cost = cost_full
207 | )
208 |
209 | # return out
210 | lst_net <- list(nodes = df_net_node, edges = df_net_edge)
211 | lst_net
212 | }
213 |
--------------------------------------------------------------------------------
/R/data.R:
--------------------------------------------------------------------------------
1 | #' Human ligand-receptor pairs
2 | #'
3 | #' Contains the names of human ligand-receptor pairs
4 | #'
5 | #' @format A data frame with two variables: \code{ligand} and \code{receptor}
6 | "lrp_human"
7 |
8 | #' Mouse ligand-receptor pairs
9 | #'
10 | #' Contains the names of mouse ligand-receptor pairs
11 | #'
12 | #' @format A data frame with two variables: \code{ligand} and \code{receptor}
13 | "lrp_mouse"
14 |
15 | #' Human protein-coding genes (PCG)
16 | #'
17 | #' Contains the names of human protein-coding genes
18 | #'
19 | #' @format A large character vector
20 | "pcg_human"
21 |
22 | #' Mouse protein-coding genes (PCG)
23 | #'
24 | #' Contains the names of mouse protein-coding genes
25 | #'
26 | #' @format A large character vector
27 | "pcg_mouse"
28 |
29 | #' Protetta Stone
30 | #'
31 | #' @format A dataframe
32 | "protetta"
33 |
34 | #' Example scRNAseq Data (CellphoneDB)
35 | #'
36 | #' @format A list
37 | "scrna_cpdb"
38 |
39 | #' Example scRNAseq Data (CytoTalk)
40 | #'
41 | #' @format A list
42 | "scrna_cyto"
43 |
44 | #' Example Pipeline Result (CytoTalk)
45 | #'
46 | #' @format A list
47 | "result_cyto"
48 |
49 | #' Graphing with Cytoscape and Graphviz
50 | #'
51 | #' @param df_net Final network; for example, the output of the
52 | #' `extract_network` function
53 | #'
54 | #' @param df_net_sub A subset of the final network (pathway); for example, the
55 | #' output of the `extract_pathways` function
56 | #'
57 | #' @param cell_type_a Name of cell type A that matches scRNA-seq file; for
58 | #' example, `"Fibroblasts"`
59 | #'
60 | #' @param dir_out Folder used for output
61 | #'
62 | #' @examples {
63 | #' pathways <- CytoTalk::result_cyto$pathways$raw
64 | #' result <- graph_pathway(pathways[[1]])
65 | #' }
66 | #'
67 | #' @return Graphs which represent the subset pathways of the final network
68 | #'
69 | #' @name doc_graphing
70 | NULL
71 | #> NULL
72 |
73 | #' Prize-Collecting Steiner Tree and Pathway Extraction
74 | #'
75 | #' @param lst_net Integrated network
76 | #'
77 | #' @param beta_max Upper limit of the test values of the PCSF objective
78 | #' function parameter $I^2$, which is inversely proportional to the total
79 | #' number of genes in a given cell-type pair; suggested to be 100 (default)
80 | #' if the total number of genes in a given cell-type pair is above 10,000; if
81 | #' the total number of genes is below 5,000, increase to 500
82 | #'
83 | #' @param omega_min Start point of omega range; omega represents the edge cost
84 | #' of the artificial network, but has been found to be less significant than
85 | #' beta. Recommended minimum of `0.5`
86 | #'
87 | #' @param omega_max End point of range between `omega_min` and `omega_max`,
88 | #' step size of `0.1`. Recommended maximum of `1.5`
89 | #'
90 | #' @param lst_pcst PCST output
91 | #'
92 | #' @param mat_pem PEM output
93 | #'
94 | #' @param beta A single beta value, see `beta_max` paramter for more detail
95 | #'
96 | #' @param omega A single omega value, see `omega_min` for more detail
97 | #'
98 | #' @param df_net Final network; for example, the output of the
99 | #' `extract_network` function
100 | #'
101 | #' @param cell_type_a Name of cell type A that matches scRNA-seq file; for
102 | #' example, `"Fibroblasts"`
103 | #'
104 | #' @param depth Starting at each ligand-receptor pair in the resultant network,
105 | #' how many steps out from that pair should be taken to generate each
106 | #' neighborhood?
107 | #'
108 | #' @examples {
109 | #' lst_net <- CytoTalk::result_cyto$integrated_net
110 | #' beta_max <- 100
111 | #' omega_min <- 0.5
112 | #' omega_max <- 0.5
113 | #' # result <- run_pcst(lst_net, beta_max, omega_min, omega_max)
114 | #' }
115 | #'
116 | #' @return Scores for the integrated network
117 | #'
118 | #' @name doc_pcst
119 | NULL
120 | #> NULL
121 |
122 | #' Integrated Co-Expression Network
123 | #'
124 | #' @param mat_type A matrix of a single cell type
125 | #'
126 | #' @param lrp A dataframe or matrix object with two columns, ligands names and
127 | #' the names of their receptors; by default, uses the `lrp_human` data. This
128 | #' package also includes `lrp_mouse`, but you can also use your own data
129 | #'
130 | #' @param mat_intra_a Intracellular network for cell type A; for example, using
131 | #' `parmigene::aracne.m` on the result of `mi_mat_parallel` will filter out
132 | #' any indirect edges per the data processing inequality
133 | #'
134 | #' @param mat_intra_b Intracellular network for cell type B
135 | #'
136 | #' @param mat_intra Intracellular network of any type
137 | #'
138 | #' @param mat_pem PEM output
139 | #'
140 | #' @param cell_type_a Name of cell type A that matches scRNA-seq file; for
141 | #' example, `"Fibroblasts"`
142 | #'
143 | #' @param cell_type_b Name of cell type B that matches scRNA-seq file; for
144 | #' example, `"LuminalEpithelialCells"`
145 | #'
146 | #' @param cell_type Name of any cell type
147 | #'
148 | #' @param vec_relev Vector containing gene relevance scores
149 | #'
150 | #' @param vec_nst_a Vector containing nonselftalk-scores for cell type A
151 |
152 | #' @param vec_nst_b Vector containing nonselftalk-scores for cell type B
153 | #'
154 | #' @examples {
155 | #' lst_scrna <- CytoTalk::scrna_cyto
156 | #' cell_type_a <- "Macrophages"
157 | #' lrp <- CytoTalk::lrp_human
158 | #' mat_a <- extract_group(cell_type_a, lst_scrna)
159 | #' result <- nonselftalk(mat_a, lrp)
160 | #' }
161 | #'
162 | #' @return Various outputs to build the integrated network
163 | #'
164 | #' @name doc_integrated
165 | NULL
166 | #> NULL
167 |
168 | #' Information Theory
169 | #'
170 | #' @param x,y Any discrete numerical vector
171 | #'
172 | #' @param mat Any discrete numerical matrix, processed column-wise
173 | #'
174 | #' @param disc Name of discretization method. Options include "equalfreq",
175 | #' "equalwidth", and "globalequalwidth". See `infotheo::discretize` for more
176 | #' detail
177 | #'
178 | #' @param nbins The number of bins by which the continuous data should be
179 | #' discretized; by default the cube root of the number of samples
180 | #'
181 | #' @param method Name of entropy estimator. Options include "emp", "mm",
182 | #' "shrink", and "sg". See `infotheo::mutinformation` for more detail
183 | #'
184 | #' @param normalize Should the entropies be normalized to fall within the range
185 | #' \[0, 1\]?
186 | #'
187 | #' @examples {
188 | #' lst_scrna <- CytoTalk::scrna_cyto
189 | #' pcg <- CytoTalk::pcg_human
190 | #' cell_type_a <- "Macrophages"
191 | #' cutoff_a <- 0.8
192 | #' mat_a <- extract_group(cell_type_a, lst_scrna)
193 | #' mat_filt_a <- subset_rownames(subset_non_zero(mat_a, cutoff_a), pcg)
194 | #' mat_disc_a <- discretize_sparse(Matrix::t(mat_filt_a))
195 | #' result <- mi_mat_parallel(mat_disc_a, method = "mm")
196 | #' }
197 | #'
198 | #' @return Mutual information calculations for filtered matrices
199 | #'
200 | #' @name doc_mutinfo
201 | NULL
202 | #> NULL
203 |
204 | #' Registering Parallel Backend
205 | #'
206 | #' @param cores How many cores to use for parallel processing?
207 | #'
208 | #' @examples {
209 | #' register_parallel(cores=2)
210 | #' unregister_parallel()
211 | #' }
212 | #'
213 | #' @return Nothing
214 | #'
215 | #' @name doc_parallel
216 | NULL
217 | #> NULL
218 |
219 | #' Preferential Expression Measure
220 | #'
221 | #' @param mat_scrna Matrix containing scRNA-seq data, along with `labels`,
222 | #' defines the transformed count data and the associated cell types
223 | #'
224 | #' @param labels Associated cell types, column-wise, of `mat_scrna`
225 | #'
226 | #' @param lst_scrna List containing scRNA-seq data; for example, lists returned
227 | #' from `read_matrix_folder` or `read_matrix_with_meta`
228 | #'
229 | #' @examples {
230 | #' lst_scrna <- CytoTalk::scrna_cyto
231 | #' result <- pem(lst_scrna)
232 | #' }
233 | #'
234 | #' @return Preferential expression measure scores
235 | #'
236 | #' @name doc_pem
237 | NULL
238 | #> NULL
239 |
--------------------------------------------------------------------------------
/R/pcst.R:
--------------------------------------------------------------------------------
1 | #' @noRd
2 | filter_arti <- function(x) {
3 | index <- startsWith(names(x), "node")
4 | x[rowSums(x[index] == "ARTI") == 0 & 0.5 <= x["omega"], ]
5 | }
6 |
7 | #' @rdname doc_pcst
8 | #' @export
9 | run_pcst <- function(lst_net, beta_max, omega_min, omega_max) {
10 | # extract dataframes
11 | df_nodes <- lst_net$nodes[, c(1, 2)]
12 | df_edges <- lst_net$edges[, c(1, 2, 3)]
13 |
14 | # generate template network
15 | df_nodes_tmp <- data.frame(node = "ARTI", prize = 1)
16 | df_edges_tmp <- data.frame(
17 | node1 = "ARTI", node2 = df_nodes$node, cost = NA)
18 |
19 | # bind to old network
20 | df_nodes_tmp <- rbind(df_nodes_tmp, df_nodes)
21 | df_edges_tmp <- rbind(df_edges, df_edges_tmp)
22 |
23 | # find index-0 node indices
24 | edge_inds <- apply(df_edges_tmp[, c(1, 2)], 2, function(x) {
25 | as.integer(match(toupper(x), toupper(df_nodes_tmp$node)) - 1)
26 | })
27 |
28 | # reticulate import (could fail)
29 | pcst_fast <- reticulate::import("pcst_fast")
30 |
31 | # for each beta
32 | lst_beta <- list()
33 | for (beta in seq_len(beta_max)) {
34 | # copy of template network
35 | df_nodes_spc <- df_nodes_tmp
36 | df_edges_spc <- df_edges_tmp
37 | index <- is.na(df_edges_tmp$cost)
38 |
39 | # beta as prize multiplier
40 | df_nodes_spc$prize <- (df_nodes_spc$prize * beta)
41 |
42 | lst_omega <- list()
43 | for (omega in seq(omega_min, omega_max, 0.1)) {
44 | # omega as ARTI edge cost
45 | df_edges_spc$cost[index] <- omega
46 |
47 | # call to Python pcst_fast function
48 | lst <- pcst_fast$pcst_fast(
49 | edge_inds,
50 | df_nodes_spc$prize,
51 | df_edges_spc$cost,
52 | as.integer(0),
53 | as.integer(1),
54 | "strong",
55 | as.integer(0)
56 | )
57 |
58 | lst$beta <- beta
59 | lst$omega <- round(omega, 1)
60 |
61 | lst_omega <- append(lst_omega, list(lst))
62 | }
63 | lst_beta <- append(lst_beta, lst_omega)
64 | }
65 |
66 | # collect the resulting lists
67 | df_nodes_all <- NULL
68 | df_edges_all <- NULL
69 | for (lst in lst_beta) {
70 | # grab run parameters
71 | beta <- lst$beta
72 | omega <- lst$omega
73 |
74 | # find node and edge names
75 | node_names <- df_nodes_tmp[lst[[1]] + 1, "node", drop=FALSE]
76 | edge_names <- df_edges_tmp[lst[[2]] + 1, c("node1", "node2")]
77 |
78 | # if there is data to add, then add it
79 | if (nrow(node_names) != 0) {
80 | df_nodes_new <- data.frame(beta, omega, node_names)
81 | df_nodes_all <- rbind(df_nodes_all, df_nodes_new)
82 | }
83 | if (nrow(edge_names) != 0) {
84 | df_edges_new <- data.frame(beta, omega, edge_names)
85 | df_edges_all <- rbind(df_edges_all, df_edges_new)
86 | }
87 | }
88 |
89 | # return out
90 | list(nodes = df_nodes_all, edges = df_edges_all)
91 | }
92 |
93 | #' @rdname doc_pcst
94 | #' @export
95 | summarize_pcst <- function(lst_pcst) {
96 | # extract dataframe,
97 | # remove artificial nodes
98 | df_edge <- filter_arti(lst_pcst[["edges"]])
99 |
100 | # count occurances, filter to less than 2000
101 | tab <- table(df_edge$beta, df_edge$omega)
102 | tab_which <- which(tab < 2000, arr.ind = TRUE, useNames = FALSE)
103 |
104 | # summary dataframe
105 | data.frame(
106 | beta = rownames(tab)[tab_which[, 1]],
107 | omega = colnames(tab)[tab_which[, 2]],
108 | n_edges = tab[tab_which]
109 | )
110 | }
111 |
112 | #' @rdname doc_pcst
113 | #' @export
114 | ks_test_pcst <- function(lst_pcst) {
115 | # extract dataframe,
116 | # remove artificial nodes
117 | df_edge <- filter_arti(lst_pcst[["edges"]])
118 |
119 | vec_param <- paste(df_edge$beta, df_edge$omega)
120 | vec_edges <- paste(df_edge$node1, df_edge$node2)
121 | tab_edges <- table(vec_edges)
122 |
123 | vec_counts <- tab_edges[cmatch(vec_edges, names(tab_edges))]
124 | lst_counts <- tapply(vec_counts, vec_param, c)
125 |
126 | # parallel loop for Kolmogorov-Smirnov test
127 | i <- NULL
128 | vec_pval <- foreach::`%dopar%`(
129 | foreach::foreach(i = seq_len(length(lst_counts)), .combine = c), {
130 | suppressWarnings(stats::ks.test(
131 | lst_counts[[i]], unlist(lst_counts[-i]),
132 | alternative = "less"
133 | )$p.value)
134 | })
135 |
136 | # order by low to hight p-values
137 | index <- order(as.numeric(vec_pval))
138 | mat_param <- do.call(rbind, strsplit(names(lst_counts)[index], " "))
139 |
140 | # test dataframe
141 | data.frame(
142 | beta = as.numeric(mat_param[, 1]),
143 | omega = as.numeric(mat_param[, 2]),
144 | pval = vec_pval[index],
145 | row.names = NULL
146 | )
147 | }
148 |
149 | #' @rdname doc_pcst
150 | #' @export
151 | extract_network <- function(lst_net, lst_pcst, mat_pem, beta, omega) {
152 | df_param <- data.frame(beta, omega)
153 | df_select <- filter_arti(merge(df_param, lst_pcst$edges))[, -c(1, 2)]
154 |
155 | node_names <- lst_net$nodes$node
156 | node_prizes <- lst_net$nodes$prize
157 |
158 | df_net <- data.frame(
159 | do.call(rbind, strsplit(df_select$node1, "__")),
160 | do.call(rbind, strsplit(df_select$node2, "__")),
161 | node_prizes[cmatch(df_select$node1, node_names)],
162 | node_prizes[cmatch(df_select$node2, node_names)],
163 | merge(df_select, lst_net$edges)$cost
164 | )
165 |
166 | names(df_net) <- c(
167 | "node1", "node1_type", "node2", "node2_type",
168 | "node1_prize", "node2_prize", "cost"
169 | )
170 |
171 | # new columns
172 | rownames(mat_pem) <- totitle(rownames(mat_pem))
173 | df_net$is_ct_edge <- (df_net$node1_type != df_net$node2_type)
174 | df_net$node1_pem <- apply(df_net, 1, function(row) {
175 | mat_pem[row["node1"], row["node1_type"]]
176 | })
177 | df_net$node2_pem <- apply(df_net, 1, function(row) {
178 | mat_pem[row["node2"], row["node2_type"]]
179 | })
180 |
181 | # reorder columns
182 | index <- c(
183 | "node1", "node2", "node1_type", "node2_type",
184 | "node1_prize", "node2_prize", "node1_pem", "node2_pem",
185 | "is_ct_edge", "cost"
186 | )
187 |
188 | # return out
189 | df_net[, index]
190 | }
191 |
192 | #' @rdname doc_pcst
193 | #' @export
194 | extract_pathways <- function(df_net, cell_type_a, depth) {
195 | # copy of dataframe
196 | df_cpy <- df_net
197 |
198 | # make names unique
199 | df_cpy$node1 <- paste0(
200 | df_cpy$node1, ifelse(df_cpy$node1_type == cell_type_a, "", "_"))
201 | df_cpy$node2 <- paste0(
202 | df_cpy$node2, ifelse(df_cpy$node2_type == cell_type_a, "", "_"))
203 |
204 | # subset to crosstalk edges
205 | df_lig <- df_cpy[df_cpy$is_ct_edge, ]
206 |
207 | if (nrow(df_lig) == 0) {
208 | return()
209 | }
210 |
211 | # loop through each
212 | df_path <- do.call(rbind, apply(df_lig, 1, function(row) {
213 | nodes_all <- unlist(df_cpy[, c(1, 2)])
214 | nodes_mat <- matrix(nodes_all, ncol = 2)
215 |
216 | nodes_sel <- c()
217 | nodes_new <- unlist(row[c(1, 2)])
218 |
219 | for (d in seq_len(depth)) {
220 | nodes_sel <- unique(c(nodes_sel, nodes_new))
221 | index <- rowSums(matrix(nodes_all %in% nodes_sel, ncol = 2)) != 0
222 | nodes_new <- as.vector(nodes_mat[index, ])
223 | }
224 |
225 | cbind(pathway = paste(row[c(1, 2)], collapse = "--"), df_cpy[index, ])
226 | }))
227 |
228 | keys <- unique(df_path$pathway)
229 | lst_path <- lapply(keys, function(x) {
230 | df_path[df_path$pathway == x, -1]
231 | })
232 | names(lst_path) <- keys
233 |
234 | # return out
235 | lst_path
236 | }
237 |
--------------------------------------------------------------------------------
/vignettes/cytotalk_guide.Rmd:
--------------------------------------------------------------------------------
1 | ---
2 | title: "CytoTalk Guide"
3 | author: "Yuxuan Hu, Shane Drabing, Kai Tan (PI)"
4 | output: rmarkdown::html_vignette
5 | vignette: >
6 | %\VignetteIndexEntry{CytoTalk Guide}
7 | %\VignetteEngine{knitr::rmarkdown}
8 | %\VignetteEncoding{UTF-8}
9 | ---
10 |
11 | ```{r, include = FALSE}
12 | knitr::opts_chunk$set(
13 | collapse = TRUE,
14 | comment = "#>"
15 | )
16 | ```
17 |
18 | ### Background
19 |
20 | Signal transduction is the primary mechanism for cell-cell communication and
21 | scRNA-seq technology holds great promise for studying this communication at
22 | high levels of resolution. Signaling pathways are highly dynamic and cross-talk
23 | among them is prevalent. Due to these two features, simply examining expression
24 | levels of ligand and receptor genes cannot reliably capture the overall
25 | activities of signaling pathways and the interactions among them.
26 |
27 |
28 |
29 |
30 |
31 | ### Prerequisites
32 |
33 | CytoTalk requires a Python module to operate correctly. To install the
34 | [`pcst_fast` module](https://github.com/fraenkel-lab/pcst_fast), please run
35 | this command *before* using CytoTalk:
36 |
37 | ``` console
38 | pip install git+https://github.com/fraenkel-lab/pcst_fast.git
39 | ```
40 |
41 | CytoTalk outputs a SIF file for use in Cytoscape. Please [install
42 | Cytoscape](https://cytoscape.org/download.html) to view the whole output
43 | network. Additionally, you'll have to install Graphviz and add the `dot`
44 | executable to your PATH. See the [Cytoscape downloads
45 | page](https://graphviz.org/download/) for more information.
46 |
47 | ### Preparation
48 |
49 | Let's assume we have a folder called "scRNAseq-data", filled with single-cell
50 | RNA sequencing datasets. Here's an example directory structure:
51 |
52 | ```{r}
53 | #> ── scRNAseq-data
54 | #> ├─ scRNAseq_BasalCells.csv
55 | #> ├─ scRNAseq_BCells.csv
56 | #> ├─ scRNAseq_EndothelialCells.csv
57 | #> ├─ scRNAseq_Fibroblasts.csv
58 | #> ├─ scRNAseq_LuminalEpithelialCells.csv
59 | #> ├─ scRNAseq_Macrophages.csv
60 | #> └─ scRNAseq_TCells.csv
61 | ```
62 |
63 |
64 |
65 | ⚠ **IMPORTANT** ⚠
66 |
67 | Notice all of these files have the prefix “scRNAseq\_” and the extension
68 | “.csv”; CytoTalk looks for files matching this pattern, so be sure to
69 | replicate it with your filenames. Let’s try reading in the folder:
70 |
71 | ``` r
72 | dir_in <- "~/Tan-Lab/scRNAseq-data"
73 | lst_scrna <- CytoTalk::read_matrix_folder(dir_in)
74 | table(lst_scrna$cell_types)
75 | ```
76 |
77 | ```{r}
78 | #> BasalCells BCells EndothelialCells
79 | #> 392 743 251
80 | #> Fibroblasts LuminalEpithelialCells Macrophages
81 | #> 700 459 186
82 | #> TCells
83 | #> 1750
84 | ```
85 |
86 | The outputted names are all the cell types we can choose to run CytoTalk
87 | against. Alternatively, we can use CellPhoneDB-style input, where one
88 | file is our data matrix, and another file maps cell types to columns
89 | (i.e. metadata):
90 |
91 | ```{r}
92 | #> ── scRNAseq-data-cpdb
93 | #> ├─ sample_counts.txt
94 | #> └─ sample_meta.txt
95 | ```
96 |
97 | There is no specific pattern required for this type of input, as both
98 | filepaths are required for the function:
99 |
100 | ``` r
101 | fpath_mat <- "~/Tan-Lab/scRNAseq-data-cpdb/sample_counts.txt"
102 | fpath_meta <- "~/Tan-Lab/scRNAseq-data-cpdb/sample_meta.txt"
103 | lst_scrna <- CytoTalk::read_matrix_with_meta(fpath_mat, fpath_meta)
104 | table(lst_scrna$cell_types)
105 | ```
106 |
107 | ```{r}
108 | #> Myeloid NKcells_0 NKcells_1 Tcells
109 | #> 1 5 3 1
110 | ```
111 |
112 | Finally, you can compose your own input list quite easily, simply have a
113 | matrix of either count or transformed data and a vector detailing the
114 | cell types of each column:
115 |
116 | ``` r
117 | mat <- matrix(rpois(90, 5), ncol = 3)
118 | cell_types <- c("TypeA", "TypeB", "TypeA")
119 | lst_scrna <- CytoTalk:::new_named_list(mat, cell_types)
120 | table(lst_scrna$cell_types)
121 | ```
122 |
123 | ```{r}
124 | #> TypeA TypeB
125 | #> 2 1
126 | ```
127 |
128 | ### Running CytoTalk
129 |
130 | Without further ado, let's run CytoTalk!
131 |
132 | ``` r
133 | # read in data folder
134 | dir_in <- "~/Tan-Lab/scRNAseq-data"
135 | lst_scrna <- CytoTalk::read_matrix_folder(dir_in)
136 |
137 | # set required parameters
138 | type_a <- "Fibroblasts"
139 | type_b <- "LuminalEpithelialCells"
140 |
141 | # run CytoTalk process
142 | results <- CytoTalk::run_cytotalk(lst_scrna, type_a, type_b)
143 | ```
144 |
145 | ```{r}
146 | #> [1 / 8] (11:15:28) Preprocessing...
147 | #> [2 / 8] (11:16:13) Mutual information matrix...
148 | #> [3 / 8] (11:20:19) Indirect edge-filtered network...
149 | #> [4 / 8] (11:20:37) Integrate network...
150 | #> [5 / 8] (11:21:44) PCSF...
151 | #> [6 / 8] (11:21:56) Determine best signaling network...
152 | #> [7 / 8] (11:21:58) Generate network output...
153 | #> [8 / 8] (11:21:59) Analyze pathways...
154 | ```
155 |
156 | All we need for a default run is the named list and selected cell types
157 | ("Macrophages" and "LuminalEpithelialCells"). The most important optional
158 | parameters to look at are `cutoff_a`, `cutoff_b`, and `beta_max`; details on
159 | these can be found in the help page for the `run_cytotalk` function (see
160 | `?run_cytotalk`). As the process runs, we see messages print to the console for
161 | each sub process.
162 |
163 | Here is what the structure of the output list looks like (abbreviated):
164 |
165 | ```{r}
166 | #> str(CytoTalk::result_cyto)
167 |
168 | #>
169 | #> List of 5
170 | #> $ params
171 | #> $ pem
172 | #> $ integrated_net
173 | #> ..$ nodes
174 | #> ..$ edges
175 | #> $ pcst
176 | #> ..$ occurances
177 | #> ..$ ks_test_pval
178 | #> ..$ final_network
179 | #> $ pathways
180 | #> ..$ raw
181 | #> ..$ graphs
182 | #> ..$ df_pval
183 | ```
184 |
185 | In the order of increasing effort, let's take a look at some of the results.
186 | Let's begin with the `results$pathways` item. This list item contains `DiagrammeR`
187 | graphs, which are viewable in RStudio, or can be exported if the `dir_out`
188 | parameter is specified during execution. Here is an example pathway
189 | neighborhood:
190 |
191 |
192 |
193 |
194 |
195 | Note that the exported SVG files (see `dir_out` parameter) are interactive,
196 | with hyperlinks to GeneCards and WikiPI. Green edges are directed from ligand
197 | to receptor. Additionally, if we specify an output directory, we can see a
198 | "cytoscape" sub-folder, which includes a SIF file read to import and two tables
199 | that can be attached to the network and used for styling. Here's an example of
200 | a styled Cytoscape network:
201 |
202 |
203 |
204 |
205 |
206 |
207 | There are a number of details we can glean from these graphs, such as node
208 | prize (side of each node), edge cost (inverse edge width), Preferential
209 | Expression Measure (intensity of each color), cell type (based on color, and
210 | shape in the Cytoscape output), and interaction type (dashed lines for
211 | crosstalk, solid for intracellular).
212 |
213 | If we want to be more formal with the pathway analysis, we can look at some
214 | scores for each neighborhood in the `results$pathways$raw` item. This list
215 | provides extracted subnetworks, based on the final network from the PCST.
216 | Additionally, the `results$pathways$df_pval` item contains a summary of the
217 | neighborhood size for each pathway, along with theoretical (Gamma distribution)
218 | test values that are found by contrsting the found pathway to random pathways
219 | from the integrated network. $p$-values for node prize, edge cost, and
220 | potential are calculated separately.
221 |
222 | ## Session Info
223 |
224 | ```{r}
225 | sessionInfo()
226 | ```
227 |
228 | ## Citing CytoTalk
229 |
230 | - Hu Y, Peng T, Gao L, Tan K. CytoTalk: *De novo* construction of signal
231 | transduction networks using single-cell transcriptomic data. ***Science
232 | Advances***, 2021, 7(16): eabf1356.
233 |
234 |
235 |
236 | - Hu Y, Peng T, Gao L, Tan K. CytoTalk: *De novo* construction of signal
237 | transduction networks using single-cell RNA-Seq data. *bioRxiv*, 2020.
238 |
239 |
240 |
241 | ## References
242 |
243 | - Shannon P, et al. Cytoscape: a software environment for integrated models of
244 | biomolecular interaction networks. *Genome Research*, 2003, 13: 2498-2504.
245 |
246 | ## Contact
247 |
248 | Kai Tan, tank1@chop.edu
249 |
250 |
251 |
--------------------------------------------------------------------------------
/R/cytotalk.R:
--------------------------------------------------------------------------------
1 | #' Main CytoTalk Pipeline
2 | #'
3 | #' @param lst_scrna List containing scRNA-seq data; for example, lists returned
4 | #' from `read_matrix_folder` or `read_matrix_with_meta`
5 | #'
6 | #' @param cell_type_a Name of cell type A that matches scRNA-seq file; for
7 | #' example, `"Fibroblasts"`
8 | #'
9 | #' @param cell_type_b Name of cell type B that matches scRNA-seq file; for
10 | #' example, `"LuminalEpithelialCells"`
11 | #'
12 | #' @param dir_out Folder used for output; if not specified, a "CytoTalk-output"
13 | #' folder will be generated
14 | #'
15 | #' @param cutoff_a Proportional threshold for lowly expressed genes in cell
16 | #' type A (range of \[0-1\]); for example, 0.2 means genes with some
17 | #' expression in at least 20% of cells are retained
18 | #'
19 | #' @param cutoff_b Proportional expression threshold for cell type B (range of
20 | #' \[0-1\])
21 | #'
22 | #' @param pcg A character vector, contains the names of protein coding genes;
23 | #' by default, uses the `pcg_human` data. This package also includes
24 | #' `pcg_mouse`, but you can also use your own data
25 | #'
26 | #' @param lrp A dataframe or matrix object with two columns, ligands names and
27 | #' the names of their receptors; by default, uses the `lrp_human` data. This
28 | #' package also includes `lrp_mouse`, but you can also use your own data
29 | #'
30 | #' @param beta_max Upper limit of the test values of the PCSF objective
31 | #' function parameter $I^2$, which is inversely proportional to the total
32 | #' number of genes in a given cell-type pair; suggested to be 100 (default)
33 | #' if the total number of genes in a given cell-type pair is above 10,000; if
34 | #' the total number of genes is below 5,000, increase to 500
35 | #'
36 | #' @param omega_min Start point of omega range; omega represents the edge cost
37 | #' of the artificial network, but has been found to be less significant than
38 | #' beta. Recommended minimum of `0.5`
39 | #'
40 | #' @param omega_max End point of range between `omega_min` and `omega_max`,
41 | #' step size of `0.1`. Recommended maximum of `1.5`
42 | #'
43 | #' @param depth Starting at each ligand-receptor pair in the resultant network,
44 | #' how many steps out from that pair should be taken to generate each
45 | #' neighborhood?
46 | #'
47 | #' @param ntrial How many random network subsets shall be created to get an
48 | #' empirical p-value for node prize and edge cost?
49 | #'
50 | #' @param cores How many cores to use for parallel processing?
51 | #'
52 | #' @param echo Should update messages be printed?
53 | #'
54 | #' @examples {
55 | #' cell_type_a <- "Macrophages"
56 | #' cell_type_b <- "LuminalEpithelialCells"
57 | #' cutoff_a <- 0.6
58 | #' cutoff_b <- 0.6
59 | #' # result <- CytoTalk::run_cytotalk(CytoTalk::scrna_cyto,
60 | #' # cell_type_a, cell_type_b,
61 | #' # cutoff_a, cutoff_b,
62 | #' # cores = 2)
63 | #' }
64 | #'
65 | #' @return A list containing model parameters, prefential expression measure,
66 | #' the integrated co-expression network, the results of the PCST, and resulting
67 | #' pathways from the final extracted network
68 | #'
69 | #' @export
70 | run_cytotalk <- function(
71 | lst_scrna, cell_type_a, cell_type_b,
72 | cutoff_a=0.2, cutoff_b=0.2,
73 | pcg=CytoTalk::pcg_human, lrp=CytoTalk::lrp_human,
74 | beta_max=100, omega_min=0.5, omega_max=0.5,
75 | depth=3, ntrial=1000,
76 | cores=NULL, echo=TRUE, dir_out=NULL) {
77 |
78 | # save numeric parameters
79 | params <- list(
80 | cell_type_a = cell_type_a, cell_type_b = cell_type_b,
81 | cutoff_a = cutoff_a, cutoff_b = cutoff_b,
82 | beta_max = beta_max, omega_min = omega_min, omega_max = omega_max,
83 | depth = depth, ntrial = ntrial
84 | )
85 |
86 | # register parallel backend
87 | if (is.null(cores) || 1 < cores) {
88 | unregister_parallel()
89 | register_parallel(cores)
90 | }
91 |
92 | # create directory
93 | if (!is.null(dir_out) && !dir.exists(dir_out)) {
94 | dir.create(dir_out, recursive = TRUE)
95 | }
96 |
97 | if (echo) {
98 | tick(1, "Preprocessing...")
99 | }
100 |
101 | mat_pem <- pem(lst_scrna)
102 | mat_a <- extract_group(cell_type_a, lst_scrna)
103 | mat_b <- extract_group(cell_type_b, lst_scrna)
104 | vec_nst_a <- nonselftalk(mat_a, lrp)
105 | vec_nst_b <- nonselftalk(mat_b, lrp)
106 | mat_filt_a <- subset_rownames(subset_non_zero_old(mat_a, cutoff_a), pcg)
107 | mat_filt_b <- subset_rownames(subset_non_zero_old(mat_b, cutoff_b), pcg)
108 |
109 | # write out PEM matrix
110 | if (!is.null(dir_out)) {
111 | fpath <- file.path(dir_out, "PEM.txt")
112 | vroom_write_silent(mat_pem, fpath, rownames = TRUE)
113 | }
114 |
115 | if (echo) {
116 | tick(2, "Mutual information matrix...")
117 | }
118 |
119 | mat_disc_a <- discretize_sparse(Matrix::t(mat_filt_a))
120 | mat_disc_b <- discretize_sparse(Matrix::t(mat_filt_b))
121 | mat_mi_a <- mi_mat_parallel(mat_disc_a, method = "mm")
122 | mat_mi_b <- mi_mat_parallel(mat_disc_b, method = "mm")
123 | dimnames(mat_mi_a) <- list(colnames(mat_disc_a), colnames(mat_disc_a))
124 | dimnames(mat_mi_b) <- list(colnames(mat_disc_b), colnames(mat_disc_b))
125 |
126 | if (echo) {
127 | tick(3, "Indirect edge-filtered network...")
128 | }
129 |
130 | mat_intra_a <- Matrix::Matrix(parmigene::aracne.m(zero_diag(mat_mi_a)))
131 | mat_intra_b <- Matrix::Matrix(parmigene::aracne.m(zero_diag(mat_mi_b)))
132 |
133 | if (echo) {
134 | tick(4, "Integrate network...")
135 | }
136 |
137 | lst_net <- integrate_network(
138 | vec_nst_a, vec_nst_b, mat_intra_a, mat_intra_b,
139 | cell_type_a, cell_type_b, mat_pem, mat_a, lrp
140 | )
141 |
142 | # write out integrated nodes and edges
143 | if (!is.null(dir_out)) {
144 | fpath <- file.path(dir_out, "IntegratedNodes.txt")
145 | vroom_write_silent(lst_net$nodes, fpath)
146 | fpath <- file.path(dir_out, "IntegratedEdges.txt")
147 | vroom_write_silent(lst_net$edges, fpath)
148 | }
149 |
150 | if (echo) {
151 | tick(5, "PCSF...")
152 | }
153 |
154 | lst_pcst <- run_pcst(lst_net, beta_max, omega_min, omega_max)
155 |
156 | # write out PCST nodes and edges
157 | if (!is.null(dir_out)) {
158 | fpath <- file.path(dir_out, "PCSTNodeOccurance.txt")
159 | vroom_write_silent(lst_pcst$nodes, fpath)
160 | fpath <- file.path(dir_out, "PCSTEdgeOccurance.txt")
161 | vroom_write_silent(lst_pcst$edges, fpath)
162 | }
163 |
164 | if (echo) {
165 | tick(6, "Determine best signaling network...")
166 | }
167 |
168 | df_test <- ks_test_pcst(lst_pcst)
169 | index <- order(as.numeric(df_test[, "pval"]))[1]
170 | beta <- df_test[index, "beta"]
171 | omega <- df_test[index, "omega"]
172 |
173 | # write out PCST scores
174 | if (!is.null(dir_out)) {
175 | vroom_write_silent(df_test, file.path(dir_out, "PCSTScores.txt"))
176 | }
177 |
178 | if (echo) {
179 | tick(7, "Generate network output...")
180 | }
181 |
182 | df_net <- extract_network(lst_net, lst_pcst, mat_pem, beta, omega)
183 | lst_path <- extract_pathways(df_net, cell_type_a, depth)
184 | lst_graph <- lapply(lst_path, graph_pathway)
185 |
186 | # no pathways found
187 | if (is.null(lst_path)) {
188 | result <- list(
189 | params = params,
190 | pem = mat_pem,
191 | integrated_net = lst_net,
192 | pcst = list(
193 | occurances = lst_pcst,
194 | ks_test_pval = df_test,
195 | final_network = df_net
196 | ),
197 | pathways = NULL
198 | )
199 |
200 | # unregister parallel backend
201 | if (is.null(cores) || 1 < cores) {
202 | unregister_parallel()
203 | }
204 |
205 | tick(8, "NOTE: No pathways found, analysis skipped!")
206 | return(result)
207 | }
208 |
209 | # write out pathways
210 | if (!is.null(dir_out)) {
211 | dir_path <- file.path(dir_out, "pathways")
212 | if (!dir.exists(dir_path)) {
213 | dir.create(dir_path, recursive = TRUE)
214 | }
215 | fnames <- names(lst_path)
216 | for (fn in fnames) {
217 | fpath <- file.path(dir_path, sprintf("%s.txt", fn))
218 | vroom_write_silent(lst_path[[fn]], fpath)
219 | }
220 |
221 | dir_gv <- file.path(dir_out, "graphviz")
222 | if (!dir.exists(dir_gv)) {
223 | dir.create(dir_gv, recursive = TRUE)
224 | }
225 | fnames <- names(lst_graph)
226 | for (fn in fnames) {
227 | fpath <- file.path(dir_gv, sprintf("%s.svg", fn))
228 | content <- DiagrammeRsvg::export_svg(lst_graph[[fn]])
229 | write(content, fpath)
230 | }
231 | }
232 |
233 | # write out final network
234 | if (!is.null(dir_out)) {
235 | write_network_sif(df_net, cell_type_a, dir_out)
236 | vroom_write_silent(df_net, file.path(dir_out, "FinalNetwork.txt"))
237 | }
238 |
239 | if (echo) {
240 | tick(8, "Analyze pathways...")
241 | }
242 |
243 | lst_pval <- lapply(
244 | lst_path, analyze_pathway, lst_net,
245 | cell_type_a, cell_type_b, beta, ntrial
246 | )
247 |
248 | # format the ligand and receptor cell types
249 | nodes <- do.call(rbind, strsplit(names(lst_path), "--"))
250 | df_pval <- do.call(rbind, apply(nodes, 1, function(x) {
251 | t <- ifelse(endsWith(x, "_"), cell_type_b, cell_type_a)
252 | x <- gsub("_$", "", x)
253 | data.frame(
254 | ligand = x[1], receptor = x[2],
255 | ligand_type = t[1], receptor_type = t[2]
256 | )
257 | }))
258 |
259 | # combine with scores and sort
260 | df_pval <- cbind(df_pval, do.call(rbind, lst_pval))
261 | df_pval <- df_pval[order(as.numeric(df_pval$pval_potential)), ]
262 |
263 | # write out analysis
264 | if (!is.null(dir_out)) {
265 | fpath <- file.path(dir_out, "PathwayAnalysis.txt")
266 | vroom_write_silent(df_pval, fpath)
267 | }
268 |
269 | # return out
270 | result <- list(
271 | params = params,
272 | pem = mat_pem,
273 | integrated_net = lst_net,
274 | pcst = list(
275 | occurances = lst_pcst,
276 | ks_test_pval = df_test,
277 | final_network = df_net
278 | ),
279 | pathways = list(
280 | raw = lst_path,
281 | graphs = lst_graph,
282 | df_pval = df_pval
283 | )
284 | )
285 |
286 | # write out session
287 | if (!is.null(dir_out)) {
288 | fpath <- file.path(dir_out, "CytoTalkSession.rda")
289 | save(result, file = fpath, version = 2)
290 | }
291 |
292 | # unregister parallel backend
293 | if (is.null(cores) || 1 < cores) {
294 | unregister_parallel()
295 | }
296 |
297 | result
298 | }
299 |
--------------------------------------------------------------------------------
/README.md:
--------------------------------------------------------------------------------
1 |
2 |
3 |
4 | # CytoTalk
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
14 |
15 | ## Table of Contents
16 |
17 | - [CytoTalk](#cytotalk)
18 | - [Table of Contents](#table-of-contents)
19 | - [Overview](#overview)
20 | - [Background](#background)
21 | - [Getting Started](#getting-started)
22 | - [Prerequisites](#prerequisites)
23 | - [Installation](#installation)
24 | - [Preparation](#preparation)
25 | - [Running CytoTalk](#running-cytotalk)
26 | - [Update Log](#update-log)
27 | - [Citing CytoTalk](#citing-cytotalk)
28 | - [References](#references)
29 | - [Contact](#contact)
30 |
31 | ## Overview
32 |
33 | We have developed the CytoTalk algorithm for *de novo* construction of a
34 | signaling network between two cell types using single-cell
35 | transcriptomics data. This signaling network is the union of multiple
36 | signaling pathways originating at ligand-receptor pairs. Our algorithm
37 | constructs an integrated network of intracellular and intercellular
38 | functional gene interactions. A prize-collecting Steiner tree (PCST)
39 | algorithm is used to extract the signaling network, based on node prize
40 | (cell-specific gene activity) and edge cost (functional interaction
41 | between two genes). The objective of the PCSF problem is to find an
42 | optimal subnetwork in the integrated network that includes genes with
43 | high levels of cell-type-specific expression and close connection to
44 | highly active ligand-receptor pairs.
45 |
46 | ### Background
47 |
48 | Signal transduction is the primary mechanism for cell-cell communication
49 | and scRNA-seq technology holds great promise for studying this
50 | communication at high levels of resolution. Signaling pathways are
51 | highly dynamic and cross-talk among them is prevalent. Due to these two
52 | features, simply examining expression levels of ligand and receptor
53 | genes cannot reliably capture the overall activities of signaling
54 | pathways and the interactions among them.
55 |
56 | ## Getting Started
57 |
58 | ### Prerequisites
59 |
60 | CytoTalk requires a Python module to operate correctly. To install the
61 | [`pcst_fast` module](https://github.com/fraenkel-lab/pcst_fast), please
62 | run this command *before* using CytoTalk:
63 |
64 | ``` console
65 | pip install git+https://github.com/fraenkel-lab/pcst_fast.git
66 | ```
67 |
68 | CytoTalk outputs a SIF file for use in Cytoscape. Please [install
69 | Cytoscape](https://cytoscape.org/download.html) to view the whole output
70 | network. Additionally, you’ll have to install Graphviz and add the `dot`
71 | executable to your PATH. See the [Cytoscape downloads
72 | page](https://graphviz.org/download/) for more information.
73 |
74 | ### Installation
75 |
76 | If you have `devtools` installed, you can use the `install_github`
77 | function directly on this repository:
78 |
79 | ``` r
80 | devtools::install_github("tanlabcode/CytoTalk")
81 | ```
82 |
83 | ### Preparation
84 |
85 | Let’s assume we have a folder called “scRNAseq-data”, filled with
86 | single-cell RNA sequencing datasets. Here’s an example directory
87 | structure:
88 |
89 | ``` txt
90 | ── scRNAseq-data
91 | ├─ scRNAseq_BasalCells.csv
92 | ├─ scRNAseq_BCells.csv
93 | ├─ scRNAseq_EndothelialCells.csv
94 | ├─ scRNAseq_Fibroblasts.csv
95 | ├─ scRNAseq_LuminalEpithelialCells.csv
96 | ├─ scRNAseq_Macrophages.csv
97 | └─ scRNAseq_TCells.csv
98 | ```
99 |
100 |
101 |
102 | ⚠ **IMPORTANT** ⚠
103 |
104 | Notice all of these files have the prefix “scRNAseq\_” and the extension
105 | “.csv”; CytoTalk looks for files matching this pattern, so be sure to
106 | replicate it with your filenames. Let’s try reading in the folder:
107 |
108 | ``` r
109 | dir_in <- "~/Tan-Lab/scRNAseq-data"
110 | lst_scrna <- CytoTalk::read_matrix_folder(dir_in)
111 | table(lst_scrna$cell_types)
112 | ```
113 |
114 | ``` console
115 | BasalCells BCells EndothelialCells
116 | 392 743 251
117 | Fibroblasts LuminalEpithelialCells Macrophages
118 | 700 459 186
119 | TCells
120 | 1750
121 | ```
122 |
123 | The outputted names are all the cell types we can choose to run CytoTalk
124 | against. Alternatively, we can use CellPhoneDB-style input, where one
125 | file is our data matrix, and another file maps cell types to columns
126 | (i.e. metadata):
127 |
128 | ``` txt
129 | ── scRNAseq-data-cpdb
130 | ├─ sample_counts.txt
131 | └─ sample_meta.txt
132 | ```
133 |
134 | There is no specific pattern required for this type of input, as both
135 | filepaths are required for the function:
136 |
137 | ``` r
138 | fpath_mat <- "~/Tan-Lab/scRNAseq-data-cpdb/sample_counts.txt"
139 | fpath_meta <- "~/Tan-Lab/scRNAseq-data-cpdb/sample_meta.txt"
140 | lst_scrna <- CytoTalk::read_matrix_with_meta(fpath_mat, fpath_meta)
141 | table(lst_scrna$cell_types)
142 | ```
143 |
144 | ``` console
145 | Myeloid NKcells_0 NKcells_1 Tcells
146 | 1 5 3 1
147 | ```
148 |
149 | If you have a `SingleCellExperiment` object with `logcounts` and
150 | `colnames` loaded onto it, you can create an input list like so:
151 |
152 | ``` r
153 | lst_scrna <- CytoTalk::from_single_cell_experiment(sce)
154 | ```
155 |
156 | Finally, you can compose your own input list quite easily, simply have a
157 | matrix of either count or transformed data and a vector detailing the
158 | cell types of each column:
159 |
160 | ``` r
161 | mat <- matrix(rpois(90, 5), ncol = 3)
162 | cell_types <- c("TypeA", "TypeB", "TypeA")
163 | lst_scrna <- CytoTalk:::new_named_list(mat, cell_types)
164 | table(lst_scrna$cell_types)
165 | ```
166 |
167 | ``` console
168 | TypeA TypeB
169 | 2 1
170 | ```
171 |
172 | ### Running CytoTalk
173 |
174 | Without further ado, let’s run CytoTalk!
175 |
176 | ``` r
177 | # read in data folder
178 | dir_in <- "~/Tan-Lab/scRNAseq-data"
179 | lst_scrna <- CytoTalk::read_matrix_folder(dir_in)
180 |
181 | # set required parameters
182 | type_a <- "Fibroblasts"
183 | type_b <- "LuminalEpithelialCells"
184 |
185 | # run CytoTalk process
186 | results <- CytoTalk::run_cytotalk(lst_scrna, type_a, type_b)
187 | ```
188 |
189 | ``` console
190 | [1 / 8] (11:15:28) Preprocessing...
191 | [2 / 8] (11:16:13) Mutual information matrix...
192 | [3 / 8] (11:20:19) Indirect edge-filtered network...
193 | [4 / 8] (11:20:37) Integrate network...
194 | [5 / 8] (11:21:44) PCSF...
195 | [6 / 8] (11:21:56) Determine best signaling network...
196 | [7 / 8] (11:21:58) Generate network output...
197 | [8 / 8] (11:21:59) Analyze pathways...
198 | ```
199 |
200 | All we need for a default run is the named list and selected cell types
201 | (“Macrophages” and “LuminalEpithelialCells”). The most important
202 | optional parameters to look at are `cutoff_a`, `cutoff_b`, and
203 | `beta_max`; details on these can be found in the help page for the
204 | `run_cytotalk` function (see `?run_cytotalk`). As the process runs, we
205 | see messages print to the console for each sub process.
206 |
207 | Here is what the structure of the output list looks like (abbreviated):
208 |
209 | ``` r
210 | str(results)
211 | ```
212 |
213 | ``` console
214 | List of 5
215 | $ params
216 | $ pem
217 | $ integrated_net
218 | ..$ nodes
219 | ..$ edges
220 | $ pcst
221 | ..$ occurances
222 | ..$ ks_test_pval
223 | ..$ final_network
224 | $ pathways
225 | ..$ raw
226 | ..$ graphs
227 | ..$ df_pval
228 | ```
229 |
230 | In the order of increasing effort, let’s take a look at some of the
231 | results. Let’s begin with the `results$pathways` item. This list item
232 | contains `DiagrammeR` graphs, which are viewable in RStudio, or can be
233 | exported if the `dir_out` parameter is specified during execution. Here
234 | is an example pathway neighborhood:
235 |
236 |
237 |
238 |
239 |
240 |
241 |
242 | Note that the exported SVG files (see `dir_out` parameter) are
243 | interactive, with hyperlinks to GeneCards and WikiPI. Green edges are
244 | directed from ligand to receptor. Additionally, if we specify an output
245 | directory, we can see a “cytoscape” sub-folder, which includes a SIF
246 | file read to import and two tables that can be attached to the network
247 | and used for styling. Here’s an example of a styled Cytoscape network:
248 |
249 |
250 |
251 |
252 |
253 |
254 |
255 |
256 |
257 | There are a number of details we can glean from these graphs, such as
258 | node prize (side of each node), edge cost (inverse edge width),
259 | Preferential Expression Measure (intensity of each color), cell type
260 | (based on color, and shape in the Cytoscape output), and interaction
261 | type (dashed lines for crosstalk, solid for intracellular).
262 |
263 | If we want to be more formal with the pathway analysis, we can look at
264 | some scores for each neighborhood in the `results$pathways$raw` item.
265 | This list provides extracted subnetworks, based on the final network
266 | from the PCST. Additionally, the `results$pathways$df_pval` item
267 | contains a summary of the neighborhood size for each pathway, along with
268 | theoretical (Gamma distribution) test values that are found by
269 | contrsting the found pathway to random pathways from the integrated
270 | network. *p*-values for node prize, edge cost, and potential are
271 | calculated separately.
272 |
273 | ## Update Log
274 |
275 | 2021-11-30: The latest release “CytoTalk\_v0.99.0” resets the versioning
276 | numbers in anticipation for submission to Bioconductor. This newest
277 | version packages functions in a modular fashion, offering more flexible
278 | input, usage, and output of the CytoTalk subroutines.
279 |
280 | 2021-10-07: The release “CytoTalk\_v4.0.0” is a completely re-written R
281 | version of the program. Approximately half of the run time as been
282 | shaved off, the program is now cross-compatible with Windows and \*NIX
283 | systems, the file space usage is down to roughly a tenth of what it was,
284 | and graphical outputs have been made easier to import or now produce
285 | portable SVG files with embedded hyperlinks.
286 |
287 | 2021-06-08: The release “CytoTalk\_v3.1.0” is a major updated R version
288 | on the basis of v3.0.3. We have added a function to generate Cytoscape
289 | files for visualization of each ligand-receptor-associated pathway
290 | extracted from the predicted signaling network between the two given
291 | cell types. For each predicted ligand-receptor pair, its associated
292 | pathway is defined as the user-specified order of the neighborhood of
293 | the ligand and receptor in the two cell types.
294 |
295 | 2021-05-31: The release “CytoTalk\_v3.0.3” is a revised R version on the
296 | basis of v3.0.2. A bug has been fixed in this version to avoid errors
297 | occurred in some special cases. We also provided a new example
298 | “RunCytoTalk\_Example\_StepByStep.R” to run the CytoTalk algorithm in a
299 | step-by-step fashion. Please download “CytoTalk\_package\_v3.0.3.zip”
300 | from the Releases page
301 | () and refer
302 | to the user manual inside the package.
303 |
304 | 2021-05-19: The release “CytoTalk\_v3.0.2” is a revised R version on the
305 | basis of v3.0.1. A bug has been fixed in this version to avoid running
306 | errors in some extreme cases. Final prediction results will be the same
307 | as v3.0.1. Please download the package from the Releases page
308 | () and refer
309 | to the user manual inside the package.
310 |
311 | 2021-05-12: The release “CytoTalk\_v3.0.1” is an R version, which is
312 | more easily and friendly to use!! Please download the package from the
313 | Releases page
314 | () and refer
315 | to the user manual inside the package.
316 |
317 | ## Citing CytoTalk
318 |
319 | - Hu Y, Peng T, Gao L, Tan K. CytoTalk: *De novo* construction of
320 | signal transduction networks using single-cell transcriptomic data.
321 | ***Science Advances***, 2021, 7(16): eabf1356.
322 |
323 |
324 |
325 | - Hu Y, Peng T, Gao L, Tan K. CytoTalk: *De novo* construction of
326 | signal transduction networks using single-cell RNA-Seq data.
327 | *bioRxiv*, 2020.
328 |
329 |
330 |
331 | ## References
332 |
333 | - Shannon P, et al. Cytoscape: a software environment for integrated
334 | models of biomolecular interaction networks. *Genome Research*,
335 | 2003, 13: 2498-2504.
336 |
337 | ## Contact
338 |
339 | Kai Tan,
340 |
341 |
342 |
--------------------------------------------------------------------------------
/README.Rmd:
--------------------------------------------------------------------------------
1 | ---
2 | output: github_document
3 | ---
4 |
5 |
6 |
7 | # CytoTalk
8 |
9 | ```{r, include = FALSE}
10 | knitr::opts_chunk$set(
11 | collapse = TRUE,
12 | comment = "#>",
13 | fig.align='center',
14 | fig.path = "man/figures/README-",
15 | out.width = "100%"
16 | )
17 | setwd(getwd())
18 | ```
19 |
20 |
21 |
22 |
23 |
24 |
25 |
26 |
27 | ## Table of Contents
28 |
29 | - [CytoTalk](#cytotalk)
30 | - [Table of Contents](#table-of-contents)
31 | - [Overview](#overview)
32 | - [Background](#background)
33 | - [Getting Started](#getting-started)
34 | - [Prerequisites](#prerequisites)
35 | - [Installation](#installation)
36 | - [Preparation](#preparation)
37 | - [Running CytoTalk](#running-cytotalk)
38 | - [Update Log](#update-log)
39 | - [Citing CytoTalk](#citing-cytotalk)
40 | - [References](#references)
41 | - [Contact](#contact)
42 |
43 | ## Overview
44 |
45 | We have developed the CytoTalk algorithm for *de novo* construction of a
46 | signaling network between two cell types using single-cell transcriptomics
47 | data. This signaling network is the union of multiple signaling pathways
48 | originating at ligand-receptor pairs. Our algorithm constructs an integrated
49 | network of intracellular and intercellular functional gene interactions. A
50 | prize-collecting Steiner tree (PCST) algorithm is used to extract the
51 | signaling network, based on node prize (cell-specific gene activity) and edge
52 | cost (functional interaction between two genes). The objective of the PCSF
53 | problem is to find an optimal subnetwork in the integrated network that
54 | includes genes with high levels of cell-type-specific expression and close
55 | connection to highly active ligand-receptor pairs.
56 |
57 | ### Background
58 |
59 | Signal transduction is the primary mechanism for cell-cell communication and
60 | scRNA-seq technology holds great promise for studying this communication at
61 | high levels of resolution. Signaling pathways are highly dynamic and cross-talk
62 | among them is prevalent. Due to these two features, simply examining expression
63 | levels of ligand and receptor genes cannot reliably capture the overall
64 | activities of signaling pathways and the interactions among them.
65 |
66 | ## Getting Started
67 |
68 | ### Prerequisites
69 |
70 | CytoTalk requires a Python module to operate correctly. To install the
71 | [`pcst_fast` module](https://github.com/fraenkel-lab/pcst_fast), please run
72 | this command *before* using CytoTalk:
73 |
74 | ``` console
75 | pip install git+https://github.com/fraenkel-lab/pcst_fast.git
76 | ```
77 |
78 | CytoTalk outputs a SIF file for use in Cytoscape. Please [install
79 | Cytoscape](https://cytoscape.org/download.html) to view the whole output
80 | network. Additionally, you'll have to install Graphviz and add the `dot`
81 | executable to your PATH. See the [Cytoscape downloads
82 | page](https://graphviz.org/download/) for more information.
83 |
84 | ### Installation
85 |
86 | If you have `devtools` installed, you can use the `install_github` function
87 | directly on this repository:
88 |
89 | ``` {r install, eval=FALSE}
90 | devtools::install_github("tanlabcode/CytoTalk")
91 | ```
92 |
93 | ### Preparation
94 |
95 | Let's assume we have a folder called "scRNAseq-data", filled with single-cell
96 | RNA sequencing datasets. Here's an example directory structure:
97 |
98 | ``` txt
99 | ── scRNAseq-data
100 | ├─ scRNAseq_BasalCells.csv
101 | ├─ scRNAseq_BCells.csv
102 | ├─ scRNAseq_EndothelialCells.csv
103 | ├─ scRNAseq_Fibroblasts.csv
104 | ├─ scRNAseq_LuminalEpithelialCells.csv
105 | ├─ scRNAseq_Macrophages.csv
106 | └─ scRNAseq_TCells.csv
107 | ```
108 |
109 |
110 |
111 | ⚠ **IMPORTANT** ⚠
112 |
113 | Notice all of these files have the prefix "scRNAseq\_" and the extension ".csv";
114 | CytoTalk looks for files matching this pattern, so be sure to replicate it
115 | with your filenames. Let's try reading in the folder:
116 |
117 | ``` r
118 | dir_in <- "~/Tan-Lab/scRNAseq-data"
119 | lst_scrna <- CytoTalk::read_matrix_folder(dir_in)
120 | table(lst_scrna$cell_types)
121 | ```
122 |
123 | ``` console
124 | BasalCells BCells EndothelialCells
125 | 392 743 251
126 | Fibroblasts LuminalEpithelialCells Macrophages
127 | 700 459 186
128 | TCells
129 | 1750
130 | ```
131 |
132 | The outputted names are all the cell types we can choose to run CytoTalk
133 | against. Alternatively, we can use CellPhoneDB-style input, where one file is
134 | our data matrix, and another file maps cell types to columns (i.e. metadata):
135 |
136 | ``` txt
137 | ── scRNAseq-data-cpdb
138 | ├─ sample_counts.txt
139 | └─ sample_meta.txt
140 | ```
141 |
142 | There is no specific pattern required for this type of input, as both filepaths
143 | are required for the function:
144 |
145 | ``` r
146 | fpath_mat <- "~/Tan-Lab/scRNAseq-data-cpdb/sample_counts.txt"
147 | fpath_meta <- "~/Tan-Lab/scRNAseq-data-cpdb/sample_meta.txt"
148 | lst_scrna <- CytoTalk::read_matrix_with_meta(fpath_mat, fpath_meta)
149 | table(lst_scrna$cell_types)
150 | ```
151 |
152 | ``` console
153 | Myeloid NKcells_0 NKcells_1 Tcells
154 | 1 5 3 1
155 | ```
156 |
157 | If you have a `SingleCellExperiment` object with `logcounts` and `colnames`
158 | loaded onto it, you can create an input list like so:
159 |
160 | ``` r
161 | lst_scrna <- CytoTalk::from_single_cell_experiment(sce)
162 | ```
163 |
164 | Finally, you can compose your own input list quite easily, simply have a matrix
165 | of either count or transformed data and a vector detailing the cell types of
166 | each column:
167 |
168 | ``` r
169 | mat <- matrix(rpois(90, 5), ncol = 3)
170 | cell_types <- c("TypeA", "TypeB", "TypeA")
171 | lst_scrna <- CytoTalk:::new_named_list(mat, cell_types)
172 | table(lst_scrna$cell_types)
173 | ```
174 |
175 | ``` console
176 | TypeA TypeB
177 | 2 1
178 | ```
179 |
180 | ### Running CytoTalk
181 |
182 | Without further ado, let's run CytoTalk!
183 |
184 | ``` r
185 | # read in data folder
186 | dir_in <- "~/Tan-Lab/scRNAseq-data"
187 | lst_scrna <- CytoTalk::read_matrix_folder(dir_in)
188 |
189 | # set required parameters
190 | type_a <- "Fibroblasts"
191 | type_b <- "LuminalEpithelialCells"
192 |
193 | # run CytoTalk process
194 | results <- CytoTalk::run_cytotalk(lst_scrna, type_a, type_b)
195 | ```
196 |
197 | ``` console
198 | [1 / 8] (11:15:28) Preprocessing...
199 | [2 / 8] (11:16:13) Mutual information matrix...
200 | [3 / 8] (11:20:19) Indirect edge-filtered network...
201 | [4 / 8] (11:20:37) Integrate network...
202 | [5 / 8] (11:21:44) PCSF...
203 | [6 / 8] (11:21:56) Determine best signaling network...
204 | [7 / 8] (11:21:58) Generate network output...
205 | [8 / 8] (11:21:59) Analyze pathways...
206 | ```
207 |
208 | All we need for a default run is the named list and selected cell types
209 | ("Macrophages" and "LuminalEpithelialCells"). The most important optional
210 | parameters to look at are `cutoff_a`, `cutoff_b`, and `beta_max`; details on
211 | these can be found in the help page for the `run_cytotalk` function (see
212 | `?run_cytotalk`). As the process runs, we see messages print to the console for
213 | each sub process.
214 |
215 | Here is what the structure of the output list looks like (abbreviated):
216 |
217 | ``` r
218 | str(results)
219 | ```
220 |
221 | ``` console
222 | List of 5
223 | $ params
224 | $ pem
225 | $ integrated_net
226 | ..$ nodes
227 | ..$ edges
228 | $ pcst
229 | ..$ occurances
230 | ..$ ks_test_pval
231 | ..$ final_network
232 | $ pathways
233 | ..$ raw
234 | ..$ graphs
235 | ..$ df_pval
236 | ```
237 |
238 | In the order of increasing effort, let's take a look at some of the results.
239 | Let's begin with the `results$pathways` item. This list item contains `DiagrammeR`
240 | graphs, which are viewable in RStudio, or can be exported if the `dir_out`
241 | parameter is specified during execution. Here is an example pathway
242 | neighborhood:
243 |
244 |
245 |
246 |
247 |
248 | Note that the exported SVG files (see `dir_out` parameter) are interactive,
249 | with hyperlinks to GeneCards and WikiPI. Green edges are directed from ligand
250 | to receptor. Additionally, if we specify an output directory, we can see a
251 | "cytoscape" sub-folder, which includes a SIF file read to import and two tables
252 | that can be attached to the network and used for styling. Here's an example of
253 | a styled Cytoscape network:
254 |
255 |
256 |
257 |
258 |
259 |
260 | There are a number of details we can glean from these graphs, such as node
261 | prize (side of each node), edge cost (inverse edge width), Preferential
262 | Expression Measure (intensity of each color), cell type (based on color, and
263 | shape in the Cytoscape output), and interaction type (dashed lines for
264 | crosstalk, solid for intracellular).
265 |
266 | If we want to be more formal with the pathway analysis, we can look at some
267 | scores for each neighborhood in the `results$pathways$raw` item. This list
268 | provides extracted subnetworks, based on the final network from the PCST.
269 | Additionally, the `results$pathways$df_pval` item contains a summary of the
270 | neighborhood size for each pathway, along with theoretical (Gamma distribution)
271 | test values that are found by contrsting the found pathway to random pathways
272 | from the integrated network. $p$-values for node prize, edge cost, and
273 | potential are calculated separately.
274 |
275 | ## Update Log
276 |
277 | 2021-11-30: The latest release "CytoTalk_v0.99.0" resets the versioning numbers
278 | in anticipation for submission to Bioconductor. This newest version packages
279 | functions in a modular fashion, offering more flexible input, usage, and output
280 | of the CytoTalk subroutines.
281 |
282 | 2021-10-07: The release "CytoTalk_v4.0.0" is a completely re-written R
283 | version of the program. Approximately half of the run time as been shaved off,
284 | the program is now cross-compatible with Windows and *NIX systems, the file
285 | space usage is down to roughly a tenth of what it was, and graphical outputs
286 | have been made easier to import or now produce portable SVG files with embedded
287 | hyperlinks.
288 |
289 | 2021-06-08: The release "CytoTalk_v3.1.0" is a major updated R version on the
290 | basis of v3.0.3. We have added a function to generate Cytoscape files for
291 | visualization of each ligand-receptor-associated pathway extracted from the
292 | predicted signaling network between the two given cell types. For each
293 | predicted ligand-receptor pair, its associated pathway is defined as the
294 | user-specified order of the neighborhood of the ligand and receptor in the two
295 | cell types.
296 |
297 | 2021-05-31: The release "CytoTalk_v3.0.3" is a revised R version on the basis
298 | of v3.0.2. A bug has been fixed in this version to avoid errors occurred in
299 | some special cases. We also provided a new example
300 | "RunCytoTalk_Example_StepByStep.R" to run the CytoTalk algorithm in a
301 | step-by-step fashion. Please download "CytoTalk_package_v3.0.3.zip" from the
302 | Releases page () and
303 | refer to the user manual inside the package.
304 |
305 | 2021-05-19: The release "CytoTalk_v3.0.2" is a revised R version on the basis
306 | of v3.0.1. A bug has been fixed in this version to avoid running errors in some
307 | extreme cases. Final prediction results will be the same as v3.0.1. Please
308 | download the package from the Releases page
309 | () and refer to the
310 | user manual inside the package.
311 |
312 | 2021-05-12: The release "CytoTalk_v3.0.1" is an R version, which is more easily
313 | and friendly to use!! Please download the package from the Releases page
314 | () and refer to the
315 | user manual inside the package.
316 |
317 | ## Citing CytoTalk
318 |
319 | - Hu Y, Peng T, Gao L, Tan K. CytoTalk: *De novo* construction of signal
320 | transduction networks using single-cell transcriptomic data. ***Science
321 | Advances***, 2021, 7(16): eabf1356.
322 |
323 |
324 |
325 | - Hu Y, Peng T, Gao L, Tan K. CytoTalk: *De novo* construction of signal
326 | transduction networks using single-cell RNA-Seq data. *bioRxiv*, 2020.
327 |
328 |
329 |
330 | ## References
331 |
332 | - Shannon P, et al. Cytoscape: a software environment for integrated models of
333 | biomolecular interaction networks. *Genome Research*, 2003, 13: 2498-2504.
334 |
335 | ## Contact
336 |
337 | Kai Tan, tank1@chop.edu
338 |
339 |
340 |
--------------------------------------------------------------------------------
/docs/pathway.svg:
--------------------------------------------------------------------------------
1 |
2 |
4 |
6 |
7 |
518 |
519 |
--------------------------------------------------------------------------------
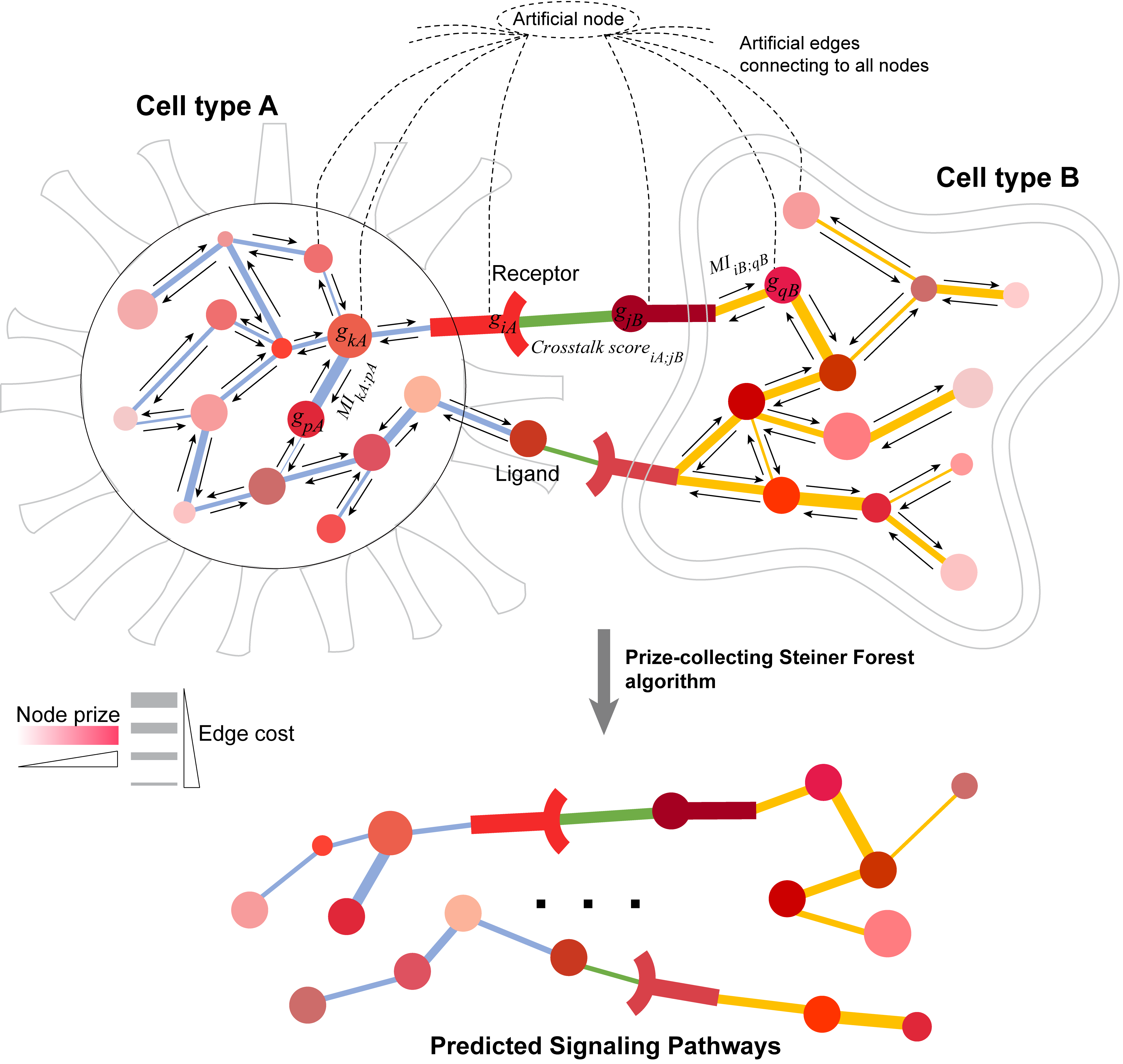 29 |
29 |  193 |
193 |  204 |
204 |  12 |
13 |
12 |
13 |  239 |
240 |
239 |
240 |  252 |
253 |
252 |
253 |